Dihydrocoumarin, an HDAC Inhibitor, Increases DNA Damage Sensitivity by Inhibiting Rad52

 , and
, and
Abstract
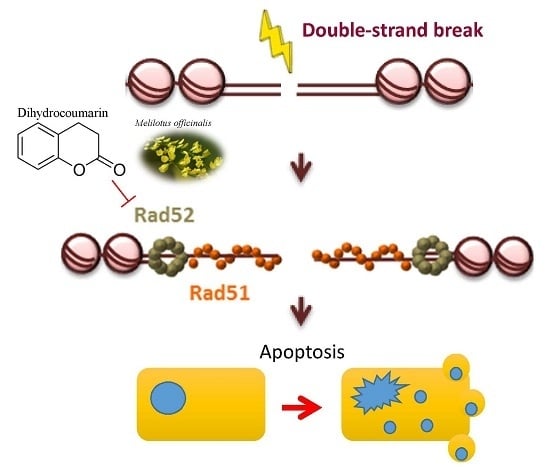
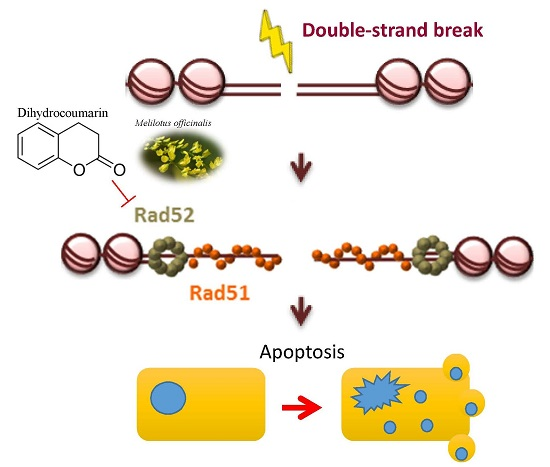
:
1. Introduction
2. Results
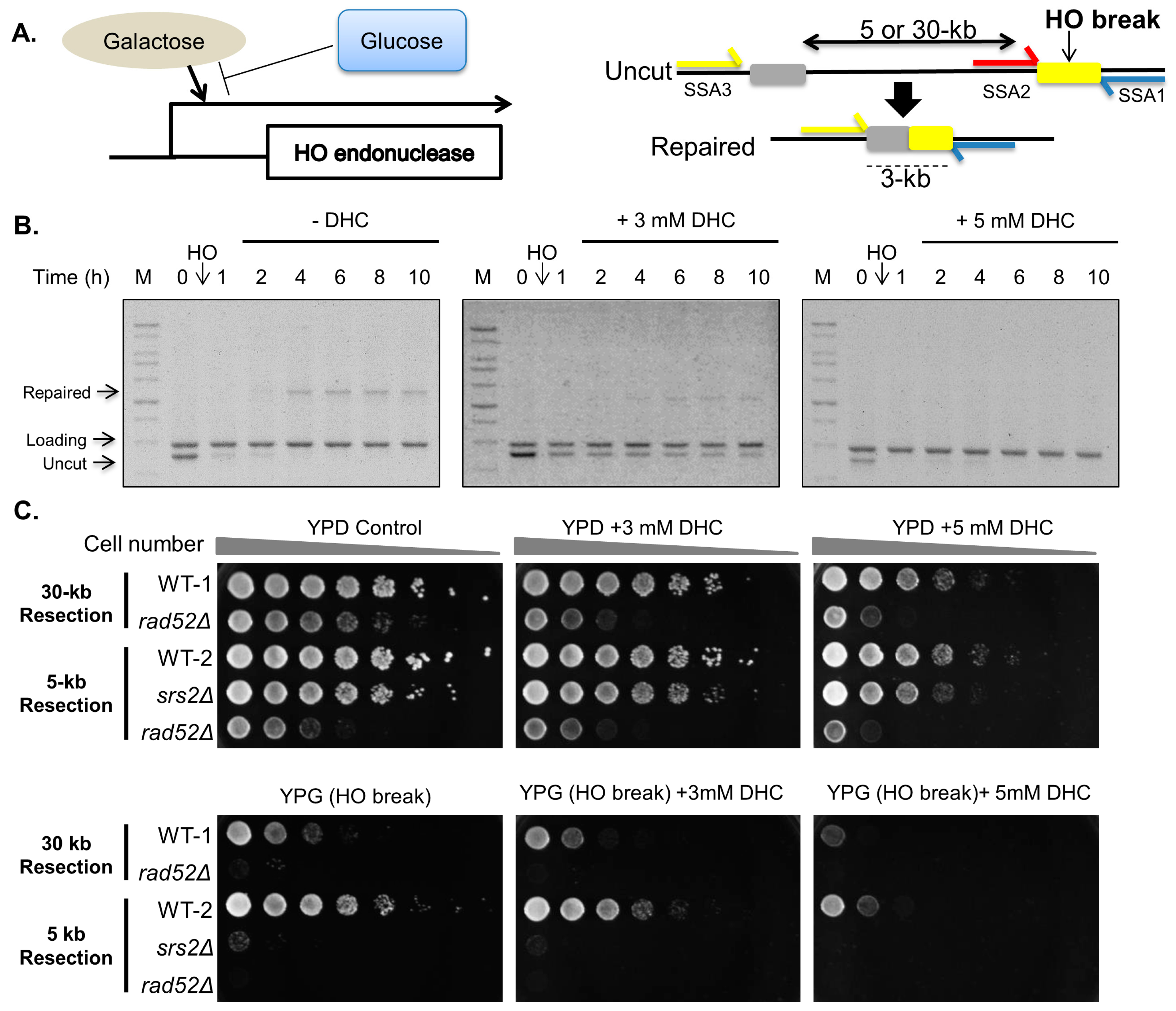
2.1. DHC (Dihydrocoumarin) Inhibits Double-Strand Break Repair
2.2. DHC Sensitizes Yeast Cells to DNA-Damaging Drugs
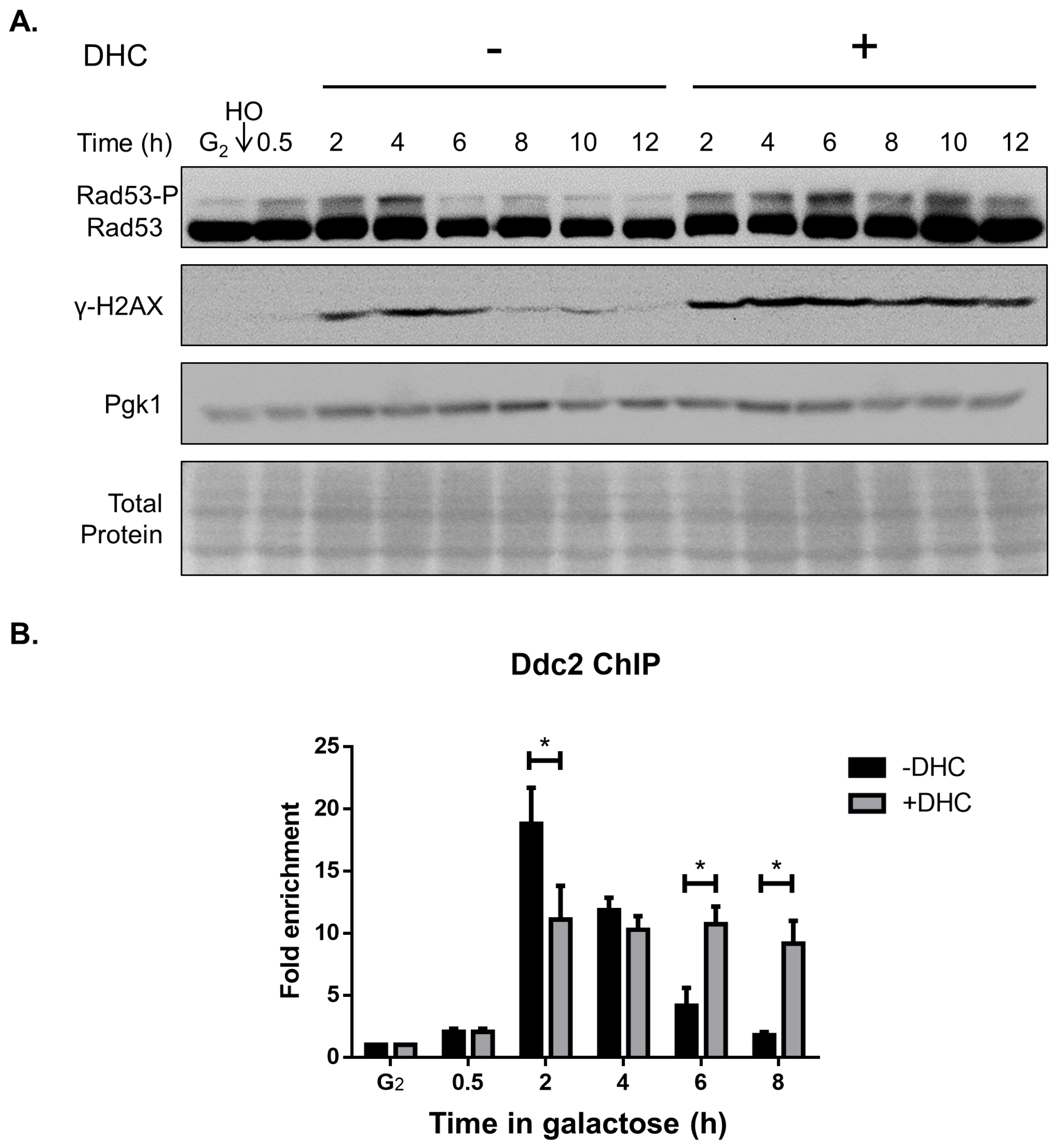
2.3. DHC Postpones DNA Damage Checkpoint Recovery
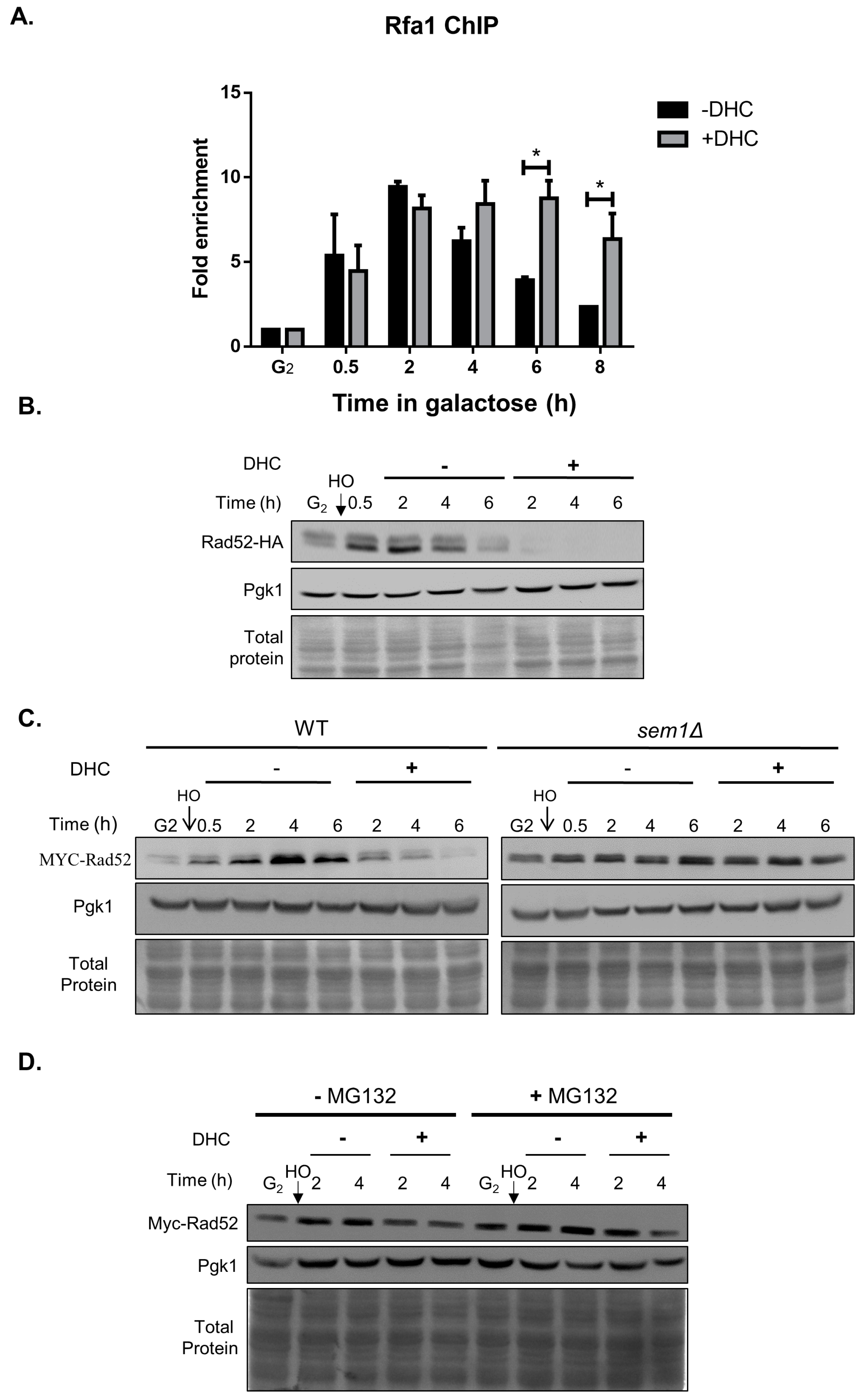
2.4. Rad52 Is Inhibited by DHC in Response to a DSB (Double-Stand Break)
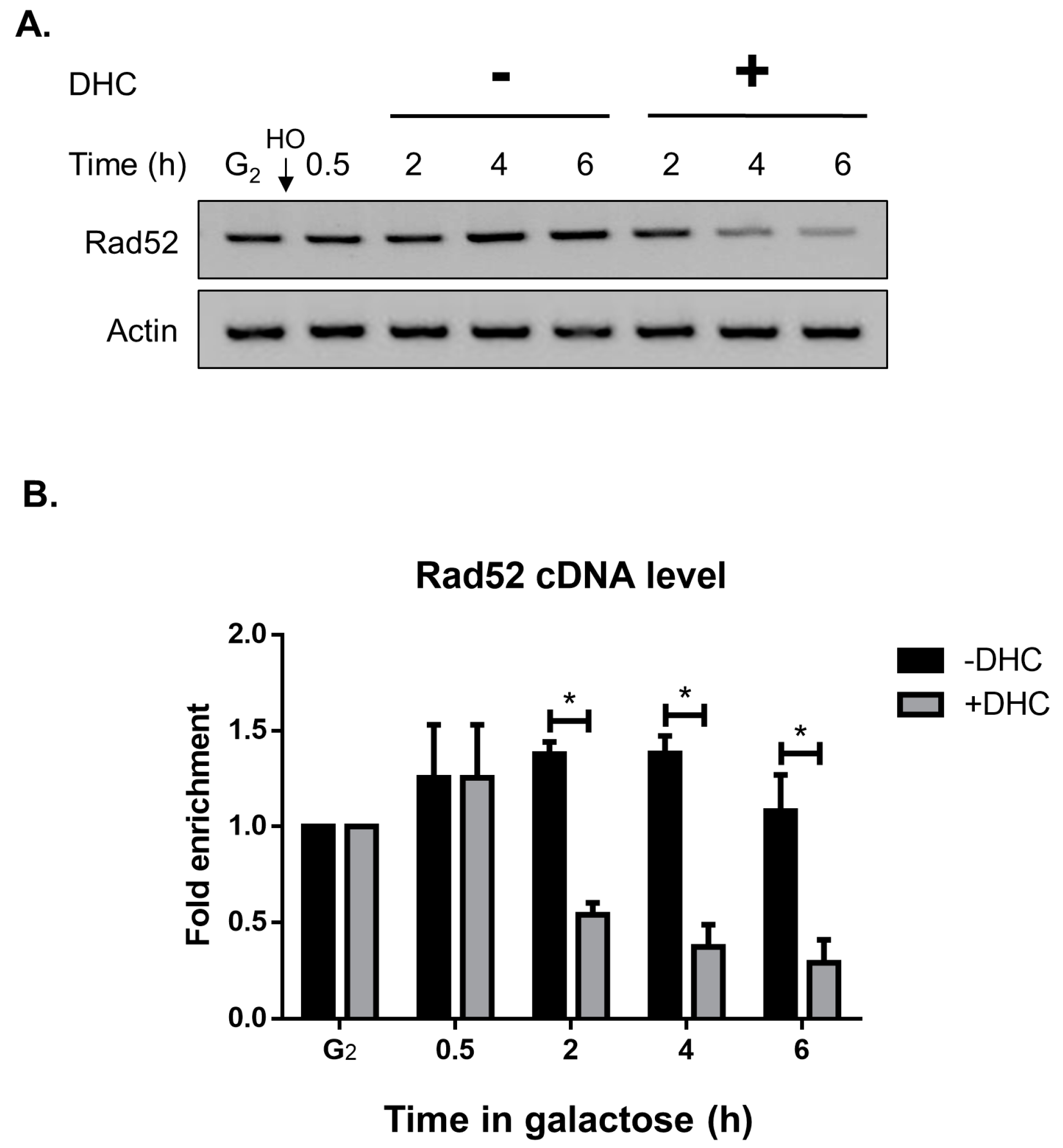
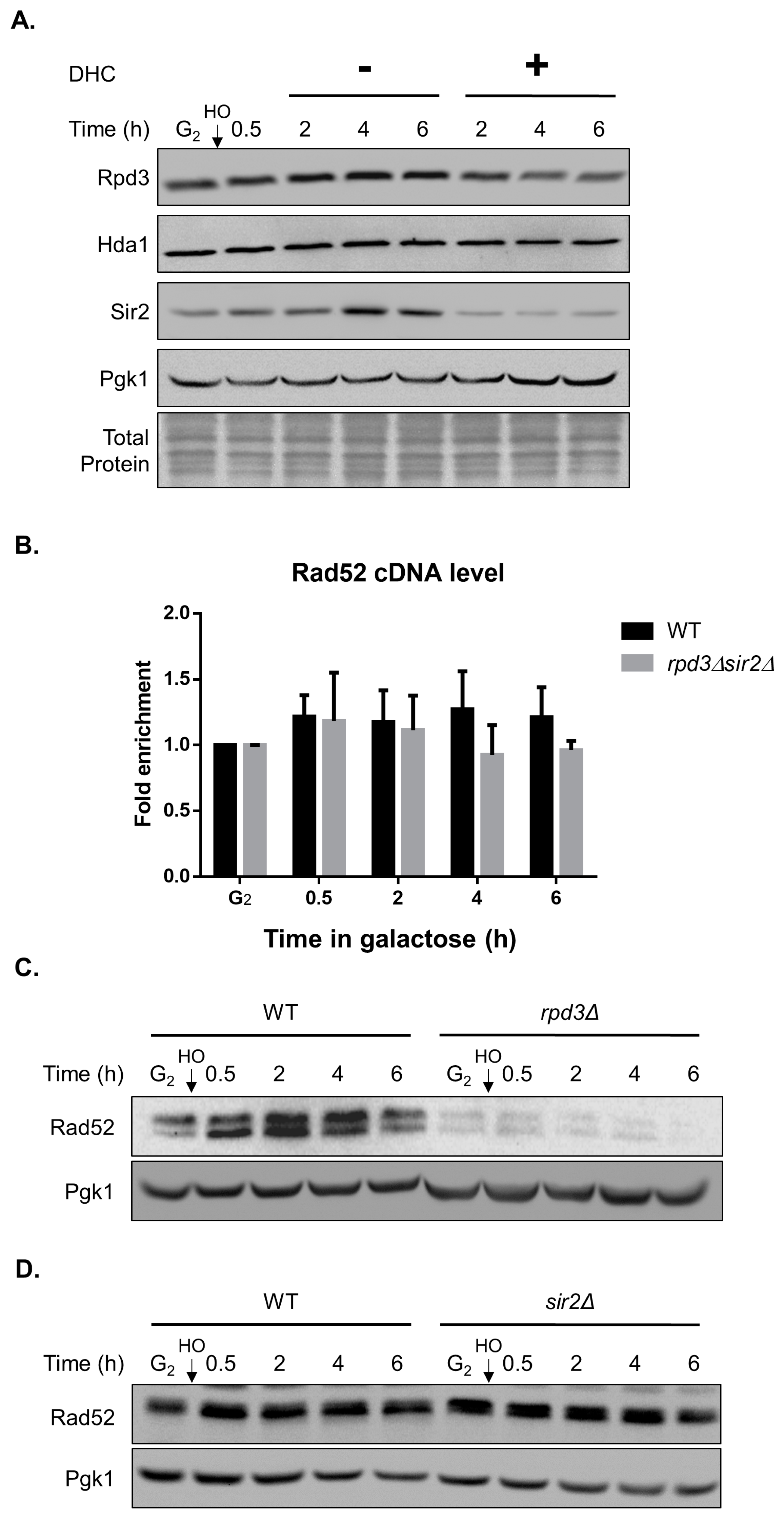
2.5. DHC Inhibits Rad52 Protein Levels through Its HDAC Inhibitor Activity
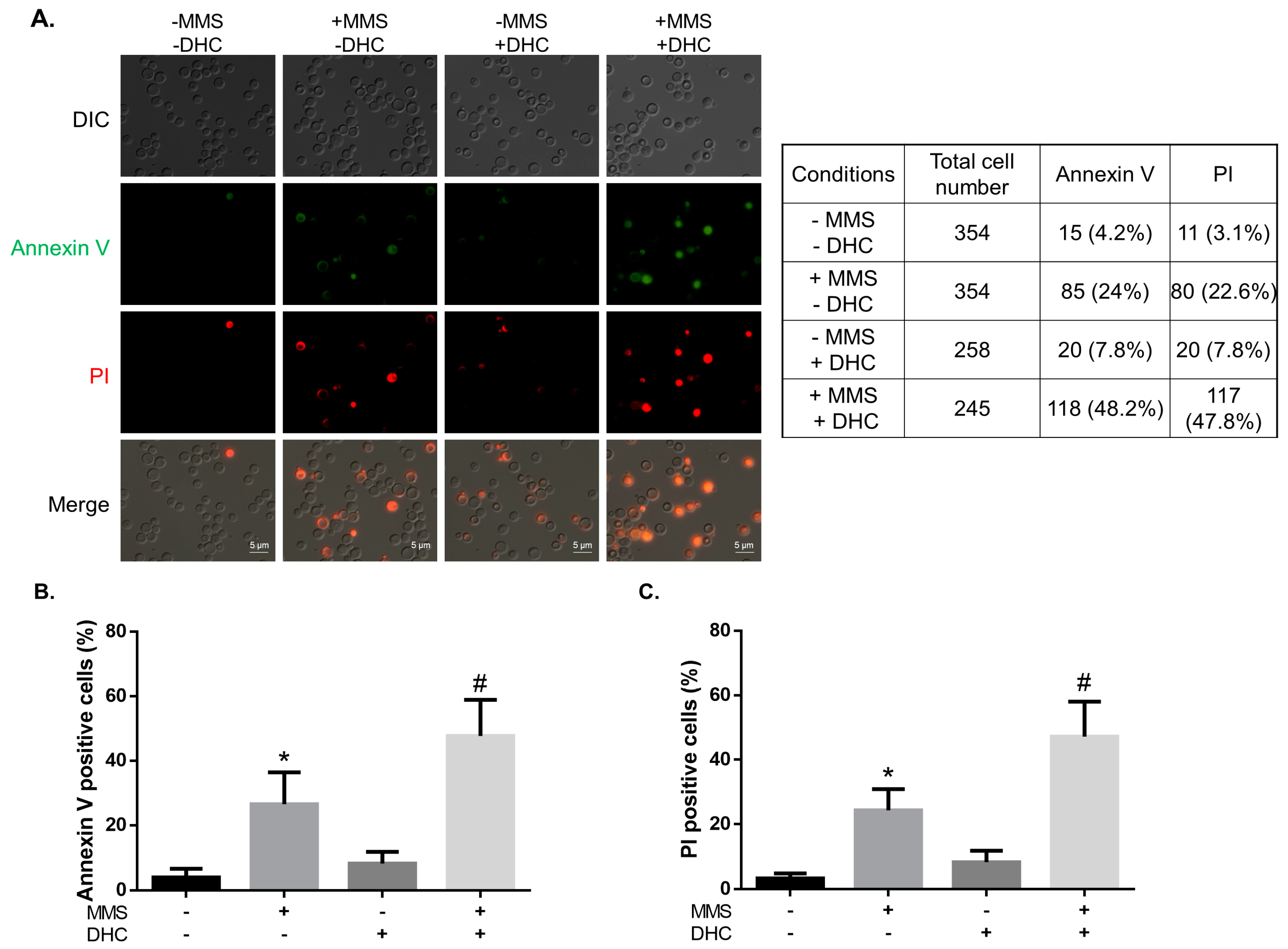
2.6. DHC Promotes Damage-Induced Apoptosis
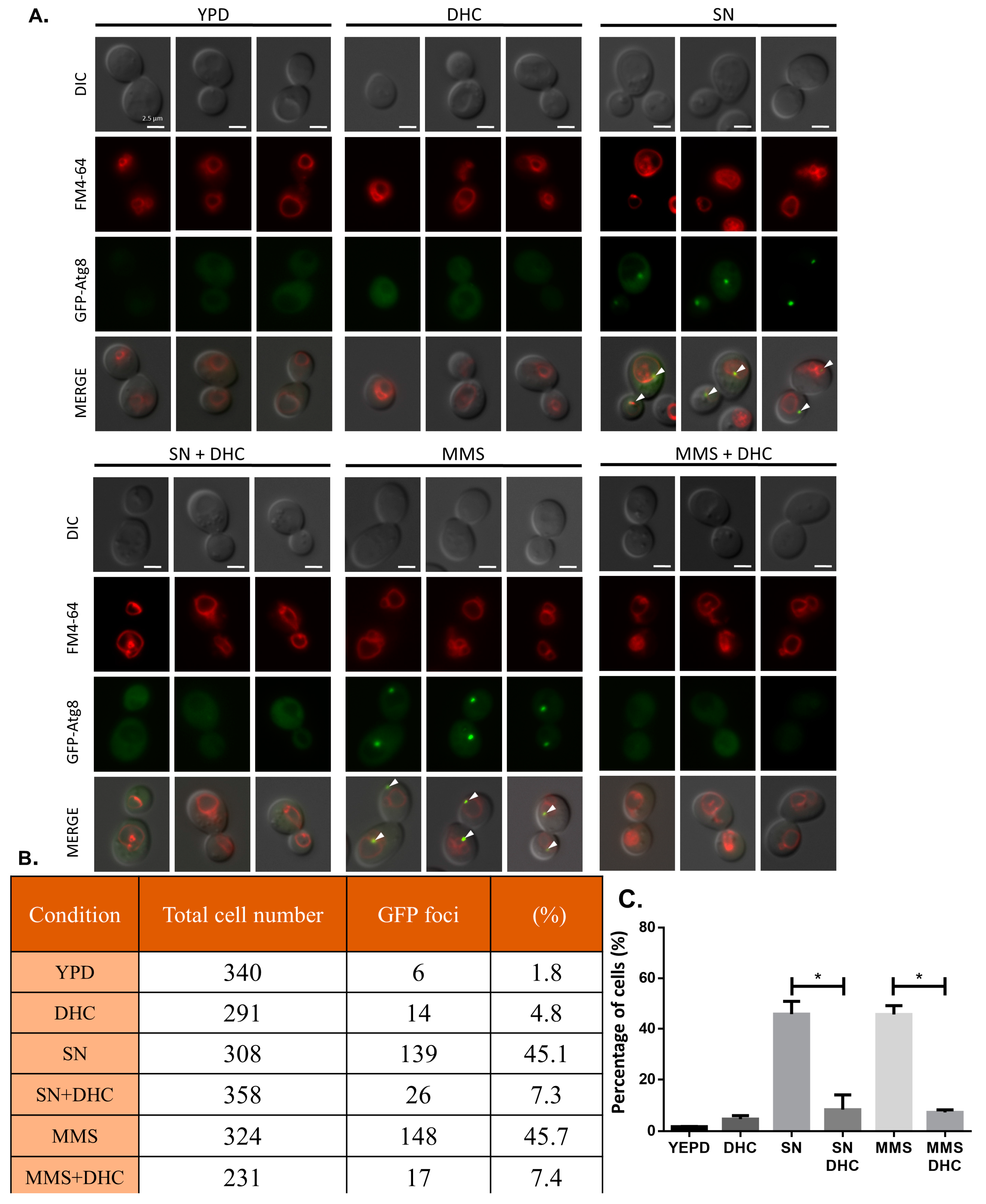
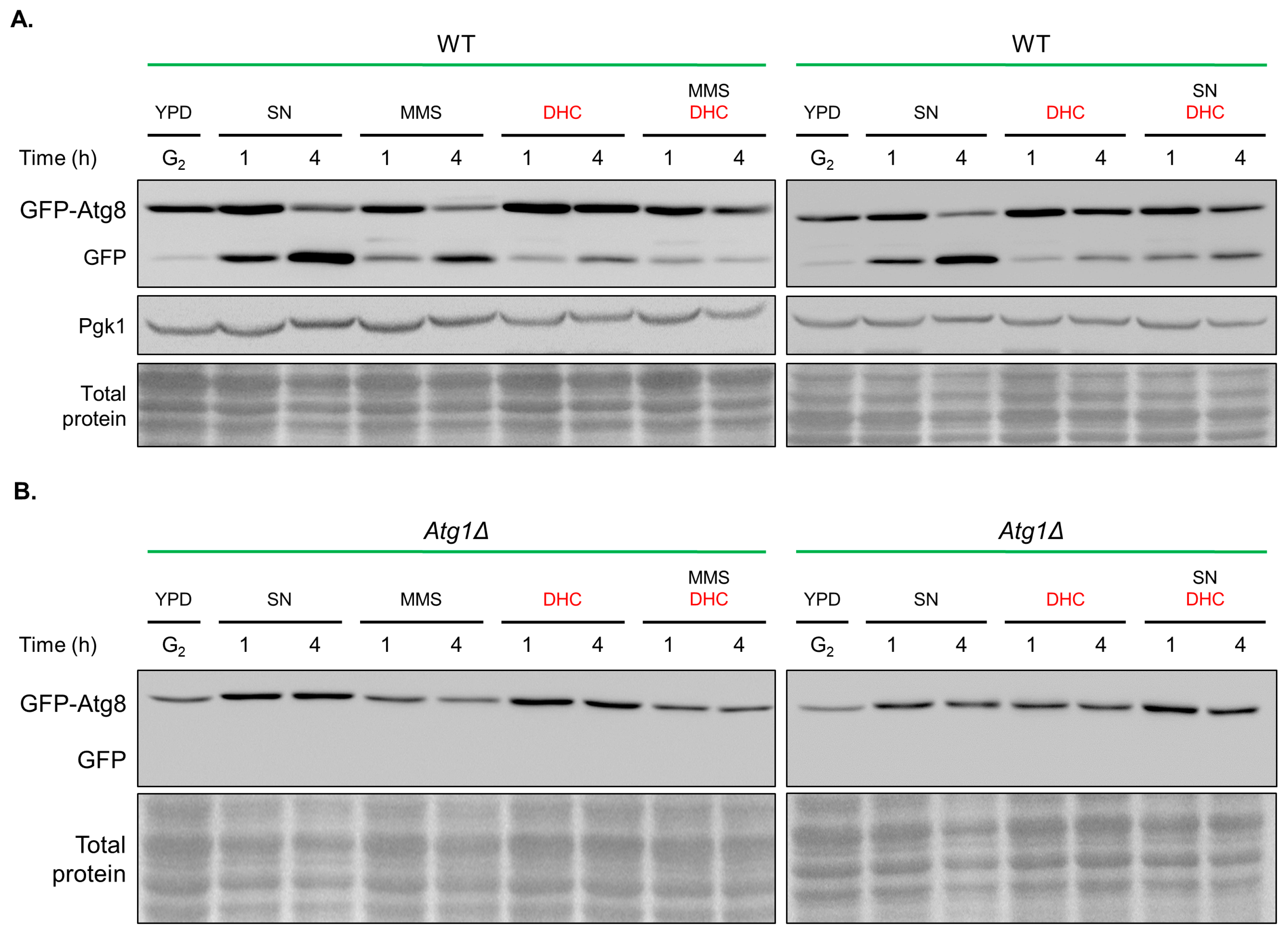
2.7. Damage-Induced Autophagy Is Impaired by DHC
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmids and Chemicals
4.2. HO Induction
4.3. Cutting and Repair Analysis
4.4. DNA Damage Sensitivity Plate Assay
4.5. Immunoblotting
4.6. Chromatin Immunoprecipitation (ChIP) Assay
4.7. RT-QPCR
4.8. Fluorescence Microscopy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhou, W.; Li, C.; Guo, M. MicroRNAs, DNA damage response, and cancer treatment. Int. J. Mol. Sci. 2016, 17, 87. [Google Scholar] [CrossRef] [PubMed]
- Swift, L.H.; Golsteyn, R.M. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int. J. Mol. Sci. 2014, 15, 3403–3431. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; van den Oord, J.J.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008, 27, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Woditschka, S.; Evans, L.; Duchnowska, R.; Reed, L.T.; Palmieri, D.; Qian, Y.; Badve, S.; Sledge, G., Jr.; Gril, B.; Aladjem, M.I.; et al. DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; McKee, C.; Muschel, R.; Haustermans, K. The effect of metformin and GANT61 combinations on the radiosensitivity of prostate cancer cells. Int. J. Mol. Sci. 2017, 18, 399. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Liu, Q. Brusatol enhances the radiosensitivity of A549 cells by promoting ROS production and enhancing DNA damage. Int. J. Mol. Sci. 2016, 17, 997. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, M.; Fujihara, H.; Fujimori, H.; Kawaguchi, K.; Yamada, H.; Nakayama, R.; Yamamoto, N.; Kishi, Y.; Hamada, Y.; Masutani, M. Synergetic effects of PARP inhibitor AZD2281 and cisplatin in oral squamous cell carcinoma in vitro and in vivo. Int. J. Mol. Sci. 2016, 17, 272. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Sloper, K.; Sorensen, C.; Syljuasen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.B.; Bartek, J.; et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar] [CrossRef] [PubMed]
- Longhese, M.P.; Foiani, M.; Muzi-Falconi, M.; Lucchini, G.; Plevani, P. DNA damage checkpoint in budding yeast. EMBO J. 1998, 17, 5525–5528. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Segurado, M.; Diffley, J.F. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008, 22, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. 2014, 26, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, S.; Di Palo, G.; Napolitano, G.; Amente, S.; Dellino, G.I.; Faretta, M.; Pelicci, P.G.; Lania, L.; Majello, B. Cell cycle-dependent resolution of DNA double-strand breaks. Oncotarget 2016, 7, 4949–4960. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Jiang, Y.; Liu, X.; Seluanov, A.; Gorbunova, V. DNA repair by homologous recombination, but not by nonhomologous end joining, is elevated in breast cancer cells. Neoplasia 2009, 11, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 2005, 280, 38631–38638. [Google Scholar] [CrossRef] [PubMed]
- Renkawitz, J.; Lademann, C.A.; Jentsch, S. Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 2014, 15, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Olaharski, A.J.; Rine, J.; Marshall, B.L.; Babiarz, J.; Zhang, L.; Verdin, E.; Smith, M.T. The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases. PLoS Genet. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lin, P.Y.; Chiu, Y.C.; Huang, J.S.; Kuo, Y.T.; Wu, J.C.; Chen, C.C. Curcumin-Mediated HDAC Inhibition Suppresses the DNA Damage response and contributes to increased DNA damage sensitivity. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Vaze, M.B.; Pellicioli, A.; Lee, S.E.; Ira, G.; Liberi, G.; Arbel-Eden, A.; Foiani, M.; Haber, J.E. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 2002, 10, 373–385. [Google Scholar] [CrossRef]
- Choy, J.S.; Kron, S.J. NuA4 subunit Yng2 function in intra-S-phase DNA damage response. Mol. Cell. Biol. 2002, 22, 8215–8225. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Bajwa, P.; Johnson, F.C.; Bhaumik, S.R.; Shilatifard, A. Rtt109 is required for proper H3K56 acetylation: A chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem. 2006, 281, 37270–37274. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, L. Molecular anatomy of the DNA damage and replication checkpoints. Radiat. Res. 2003, 159, 139–148. [Google Scholar] [CrossRef]
- Bandhu, A.; Kang, J.; Fukunaga, K.; Goto, G.; Sugimoto, K. Ddc2 mediates Mec1 activation through a Ddc1- or Dpb11-independent mechanism. PLoS Genet 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Aracely Garcia, G.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Nair, U.; Thumm, M.; Klionsky, D.J.; Krick, R. GFP-Atg8 protease protection as a tool to monitor autophagosome biogenesis. Autophagy 2011, 7, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Huang, W.P.; Stromhaug, P.E.; Klionsky, D.J. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev. Cell 2002, 3, 825–837. [Google Scholar] [CrossRef]
- Mizushima, N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. 2009, 452, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 2007, 27, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Weinberger, M.; Marchetti, M.A.; Ramachandran, L.; D’Urso, G.; Huberman, J.A. Apoptosis-like yeast cell death in response to DNA damage and replication defects. Mutat. Res. 2003, 532, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Georgieva, B.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef]
- Jin, C.; Reed, J.C. Yeast and apoptosis. Nat. Rev. Mol. Cell Biol. 2002, 3, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Eisenberg, T.; Buttner, S.; Meisinger, C.; Kroemer, G.; Madeo, F. Apoptosis in yeast: Triggers, pathways, subroutines. Cell Death Differ. 2010, 17, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Dotiwala, F.; Eapen, V.V.; Harrison, J.C.; Arbel-Eden, A.; Ranade, V.; Yoshida, S.; Haber, J.E. DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc. Natl. Acad. Sci. USA 2013, 110, E41–E49. [Google Scholar] [CrossRef] [PubMed]
- Eapen, V.V.; Waterman, D.P.; Bernard, A.; Schiffmann, N.; Sayas, E.; Kamber, R.; Lemos, B.; Memisoglu, G.; Ang, J.; Mazella, A.; et al. A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E115–E1167. [Google Scholar] [CrossRef] [PubMed]
- Pellicioli, A.; Lucca, C.; Liberi, G.; Marini, F.; Lopes, M.; Plevani, P.; Romano, A.; Di Fiore, P.P.; Foiani, M. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 1999, 18, 6561–6572. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008, 134, 231–243. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Source |
|---|---|---|
| YMV002 | MATα ho hml∆::ADE1 mata∆::ADE1 his4::-URA3-leu2-(Xho1 to Asp718)-his4 leu2::HOcs ade3::GAL::HO ade1 lys5 ura3-52 | James Haber |
| YMV037 | MATα ho hml∆::ADE1 mata∆::ADE1 his4::-URA3-leu2-(Xho1 to Asp718)-his4 leu2::HOcs ade3::GAL::HO ade1 lys5 ura3-52 rad52∆::HPH | James Haber |
| YMV045 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2 (Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 | James Haber |
| YMV046 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ADE1 leu2:HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 rad52∆::HPH (hygro) | James Haber |
| YMV 057 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2 (Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 srs2::HPH | James Haber |
| BY4741 | MATa his3∆ leu2∆ met15∆ ura3∆ | This study |
| BY4741-atg1 | MATa his3∆ leu2∆ met15∆ ura3∆ atg1∆::KAN | This study |
| BY4741-atg8 | MATa his3∆ leu2∆ met15∆ ura3∆ atg8∆::KAN | This study |
| BY4741-gcn5 | MATa his3∆ leu2∆ met15∆ ura3∆ gcn5∆::KAN | This study |
| BY4741-hat1 | MATa his3∆ leu2∆ met15∆ ura3∆ hat1∆::KAN | This study |
| BY4741-hda1 | MATa his3∆ leu2∆ met15∆ ura3∆ hda1∆::KAN | This study |
| BY4741-rad6 | MATa his3∆ leu2∆ met15∆ ura3∆ rad6∆::KAN | This study |
| BY4741-rpd3 | MATa his3∆ leu2∆ met15∆ ura3∆ rpd3∆::KAN | This study |
| BY4741-sir2 | MATa his3∆ leu2∆ met15∆ ura3∆ sir2∆::KAN | This study |
| BY4741-rtt109 | MATa his3∆ leu2∆ met15∆ ura3∆ rtt109∆::KAN | This study |
| RLY001 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2 (Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) DDC2-MYC::KAN | This study |
| YAY012 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2(Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) rpd3∆::KAN | This study |
| YAY013 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2 (Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) HA-RAD52::KAN | This study |
| YAY016 | ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2(Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG?) HA-Rad52::KAN Rpd3::TRP | This study |
| YAY028 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2(Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) MYC-RAD52::TRP | This study |
| NKY001 | ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2(Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG?) Myc-Rad52::TRP Sir2∆::KAN | This study |
| RLY004 | BY4741-atg8 transformed with PRS416 GFP-Atg8 in URA drop media | This study |
| RLY005 | BY4741-atg1 transformed with PRS416 GFP-Atg8 in URA drop media | This study |
| RLY006 | MATαho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2(Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) MYC-RAD52::TRP sem1::KAN | This study |
| DHY001 | MATα ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2(Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG?) Sir2::KAN | This study |
| Primer | Sequence |
|---|---|
| SSA1 | CCGCTGAACATACCACGTTG |
| SSA2 | CACTTCCAGATGAGGCGCTG |
| SSA3 | TGAACTCTGGTGTCTTTTAG |
| RAD3A | GATAAGATTGCGACAAAAGAGGATA |
| RAD3D | GTGGGACGAGACGTTTAGATAGTAA |
| HO-F | CCAAATCTGATGGAAGAATGGG |
| HO-R | CCGCTGAACATACCACGTTG |
| SMC2-F | ATCACTGATTGAAGAGGCAGC |
| SMC2-R | TACGAGTCTCACCGTTCTCCA |
| Rad52-int-RNA-F | TGGCTGGTCTACGGAGGTAA |
| Rad52-int-RNA-R | GCGGTGGTCATCGTTTTGTC |
| Rad52-int-QPCR-F | TCAAGTACCGCGTGAAACCA |
| Rad52-int-QPCR-R | CGATCTTTGTTGCGGAACGG |
| Actin-int-F | TACGTTTCCATCCAAGCCGT |
| Actin-int-R | CGGCAGATTCCAAACCCAAA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Huang, J.-S.; Wang, T.-H.; Kuo, C.-H.; Wang, C.-J.; Wang, S.-H.; Leu, Y.-L. Dihydrocoumarin, an HDAC Inhibitor, Increases DNA Damage Sensitivity by Inhibiting Rad52. Int. J. Mol. Sci. 2017, 18, 2655. https://doi.org/10.3390/ijms18122655
Chen C-C, Huang J-S, Wang T-H, Kuo C-H, Wang C-J, Wang S-H, Leu Y-L. Dihydrocoumarin, an HDAC Inhibitor, Increases DNA Damage Sensitivity by Inhibiting Rad52. International Journal of Molecular Sciences. 2017; 18(12):2655. https://doi.org/10.3390/ijms18122655
Chicago/Turabian StyleChen, Chin-Chuan, Ju-Sui Huang, Tong-Hong Wang, Chen-Hsin Kuo, Chia-Jen Wang, Shu-Huei Wang, and Yann-Lii Leu. 2017. "Dihydrocoumarin, an HDAC Inhibitor, Increases DNA Damage Sensitivity by Inhibiting Rad52" International Journal of Molecular Sciences 18, no. 12: 2655. https://doi.org/10.3390/ijms18122655
APA StyleChen, C. -C., Huang, J. -S., Wang, T. -H., Kuo, C. -H., Wang, C. -J., Wang, S. -H., & Leu, Y. -L. (2017). Dihydrocoumarin, an HDAC Inhibitor, Increases DNA Damage Sensitivity by Inhibiting Rad52. International Journal of Molecular Sciences, 18(12), 2655. https://doi.org/10.3390/ijms18122655