Evidence of Altered Glycosylation of Serum Proteins Prior to Pancreatic Cancer Diagnosis


 , ,
, ,
Abstract
:
1. Introduction
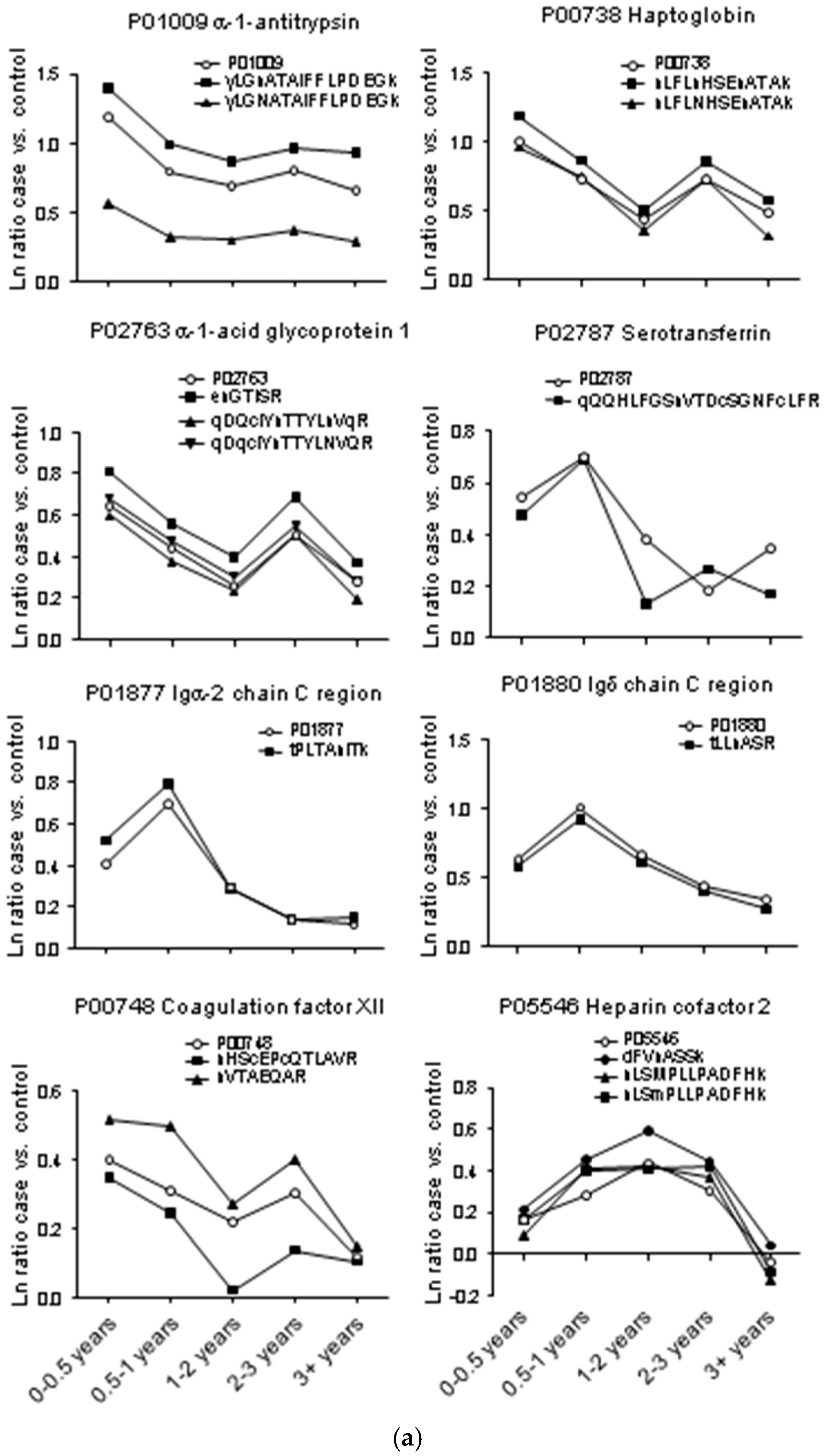
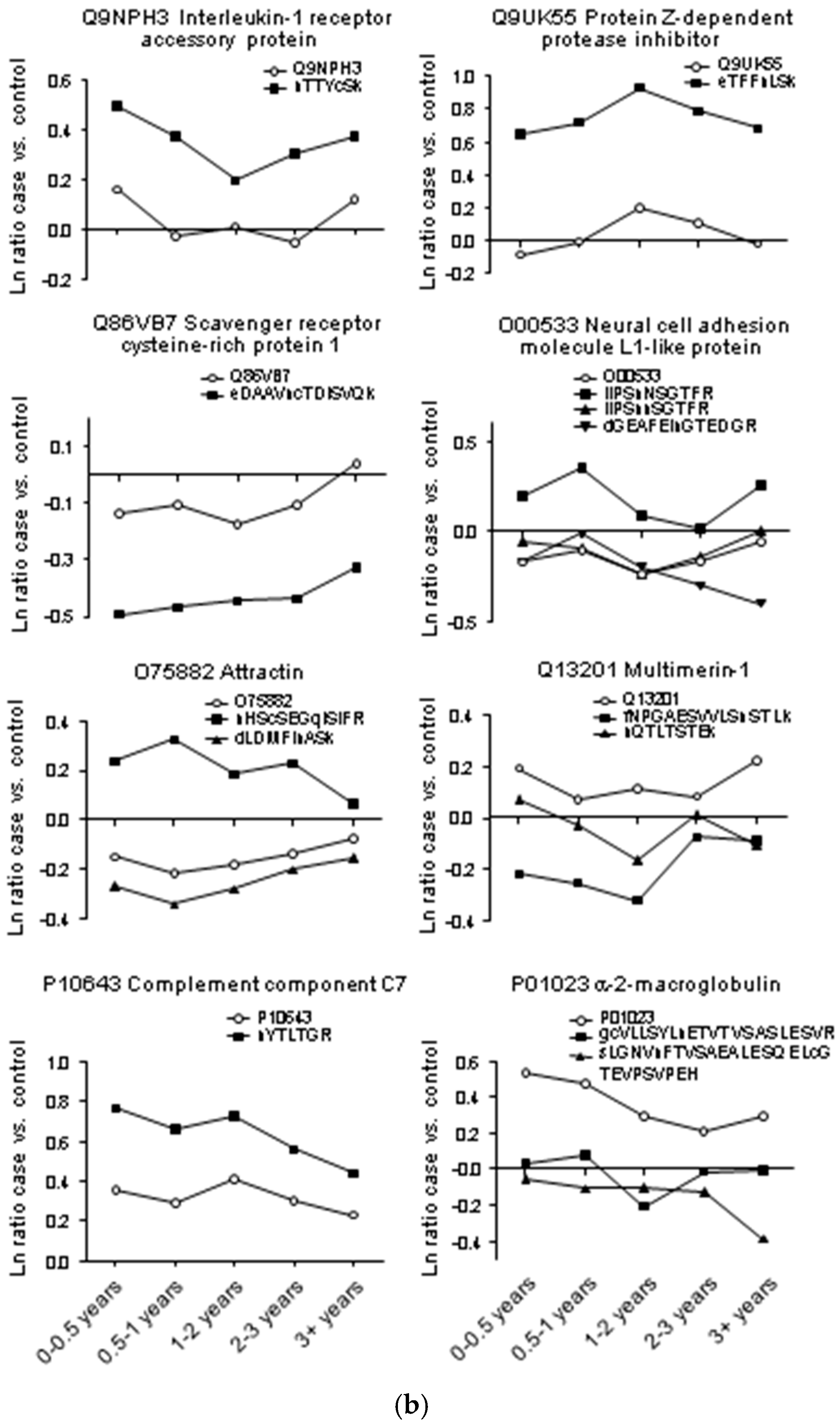
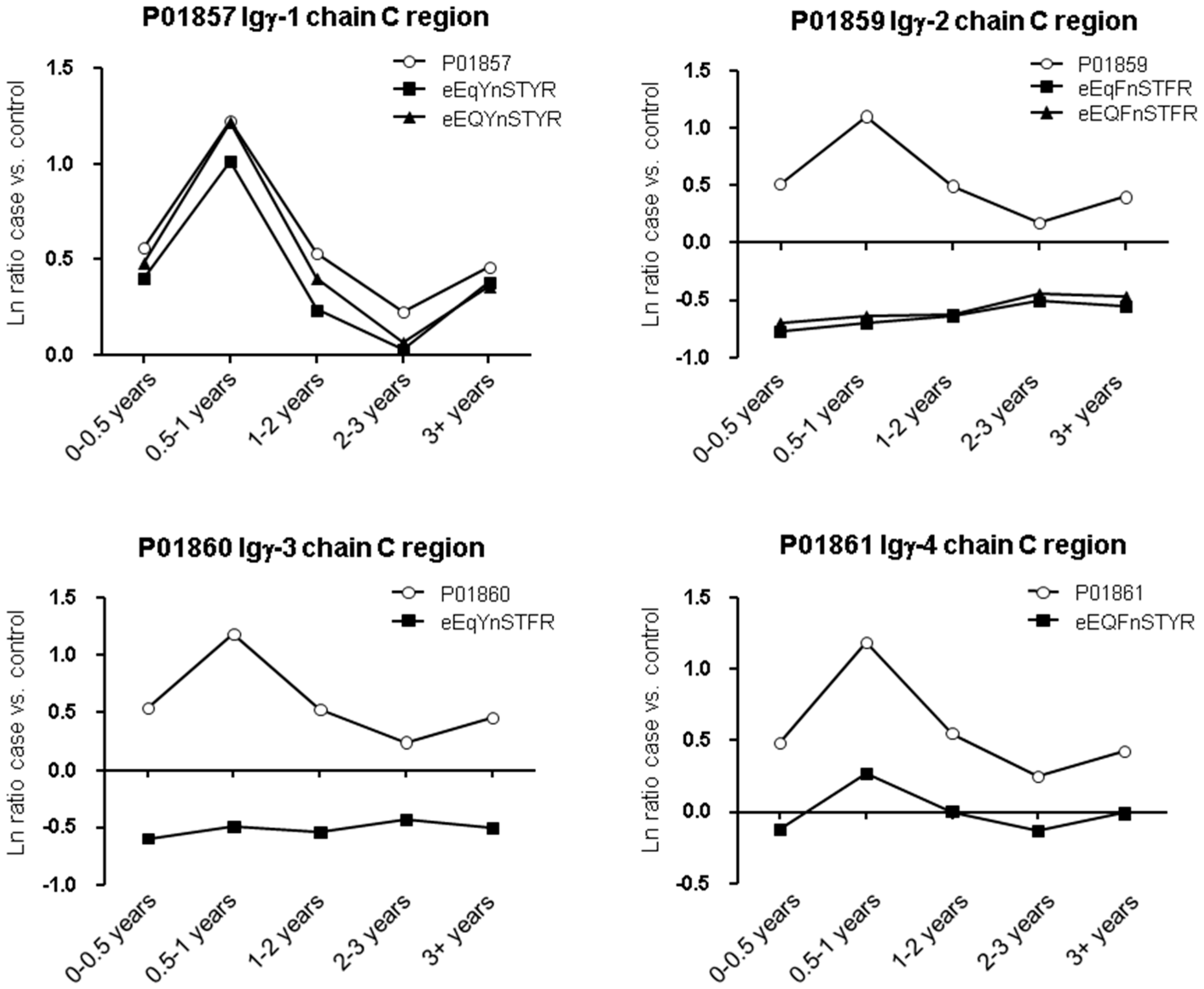
2. Results
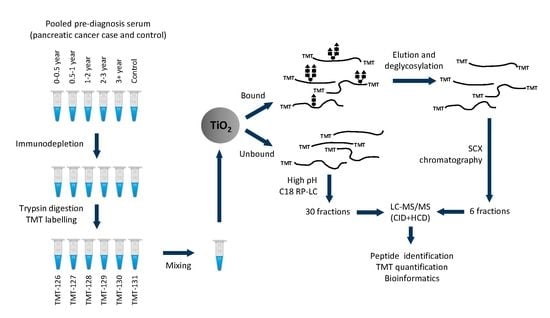
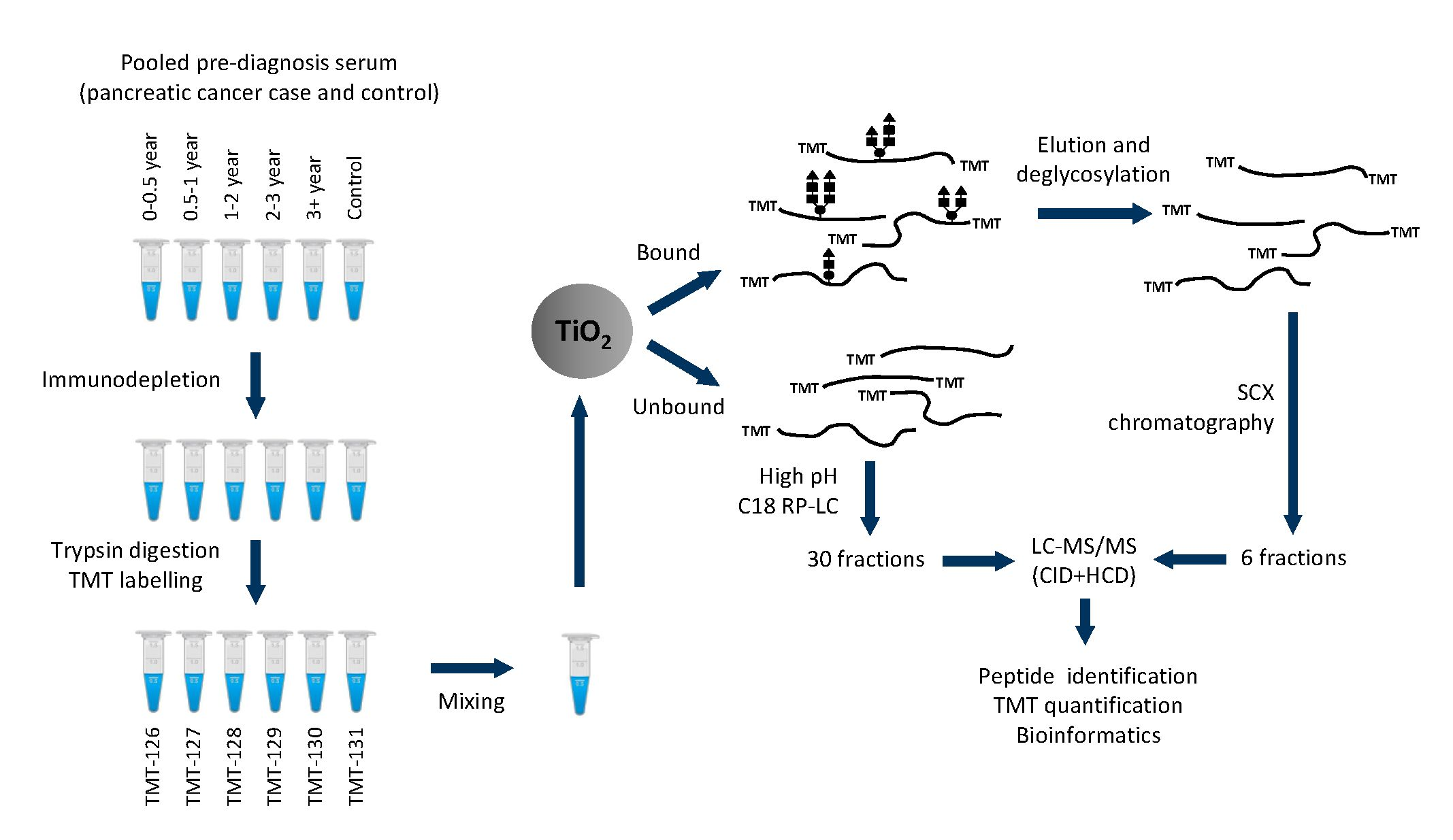
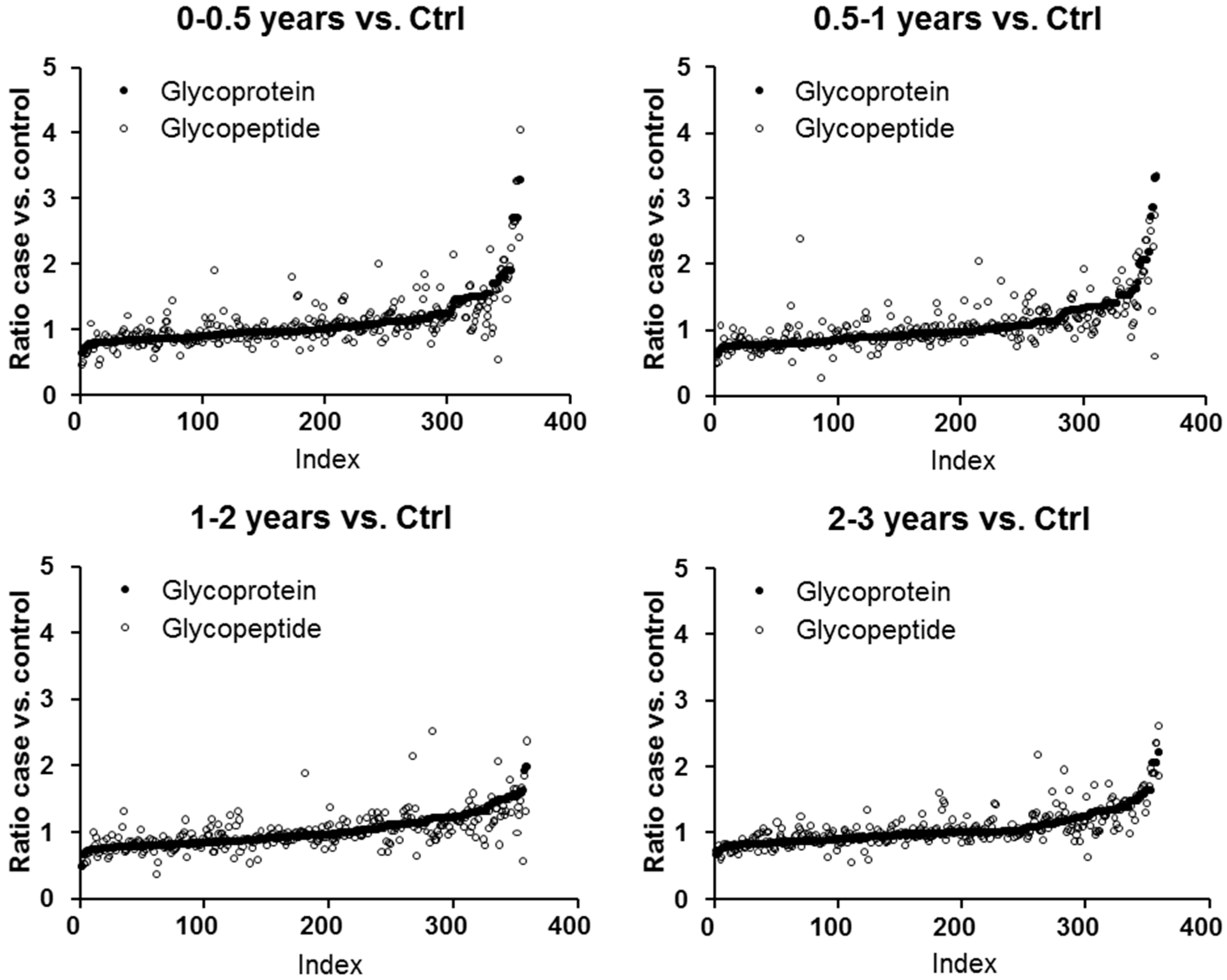
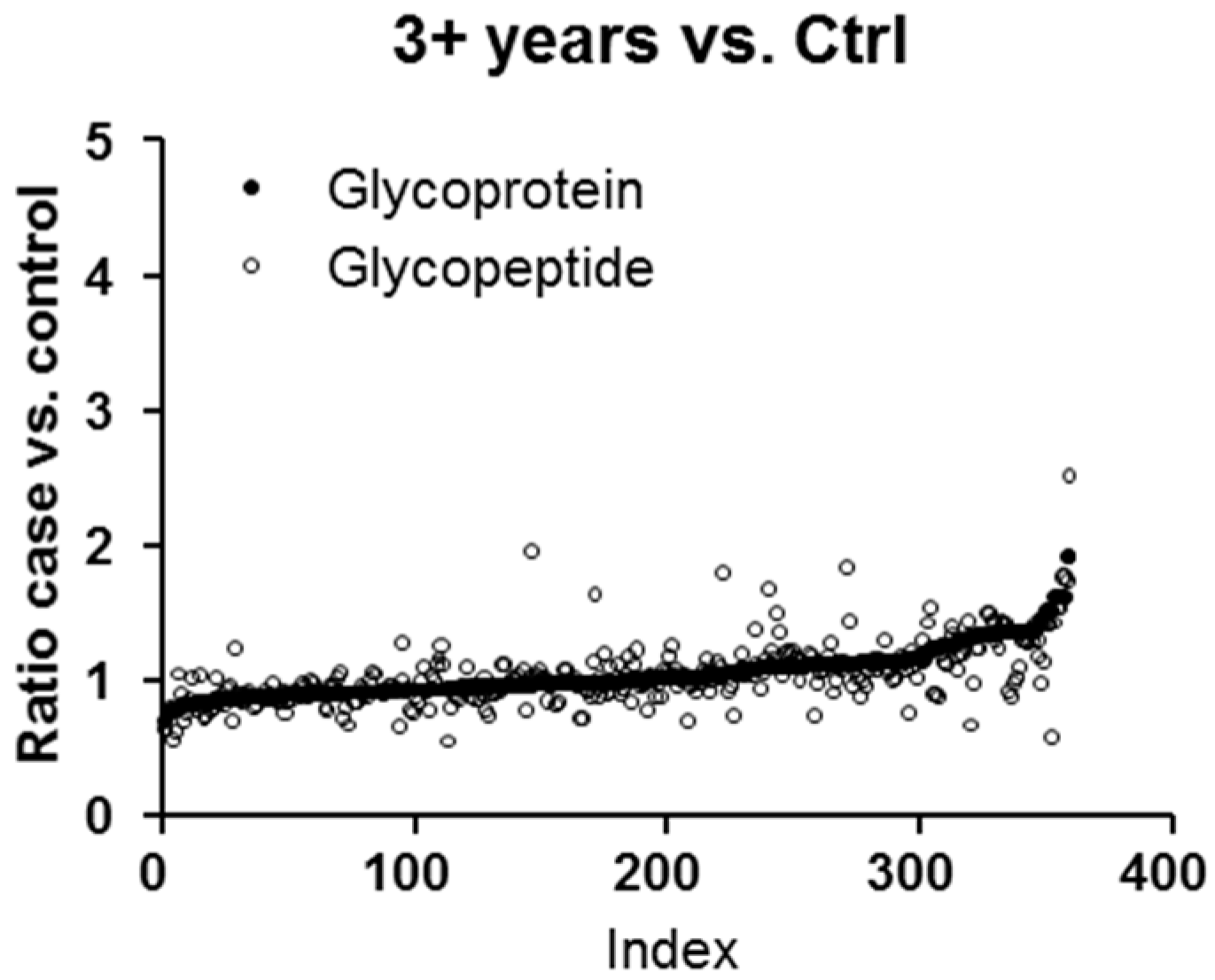
2.1. Protein and Glycopeptide Profiling of Serum from Cancer-Free Controls and Cases of Pancreatic Ductal Adenocarcinoma Taken Months to Years before Diagnosis
2.2. Identification of N-Glycosylation Sites
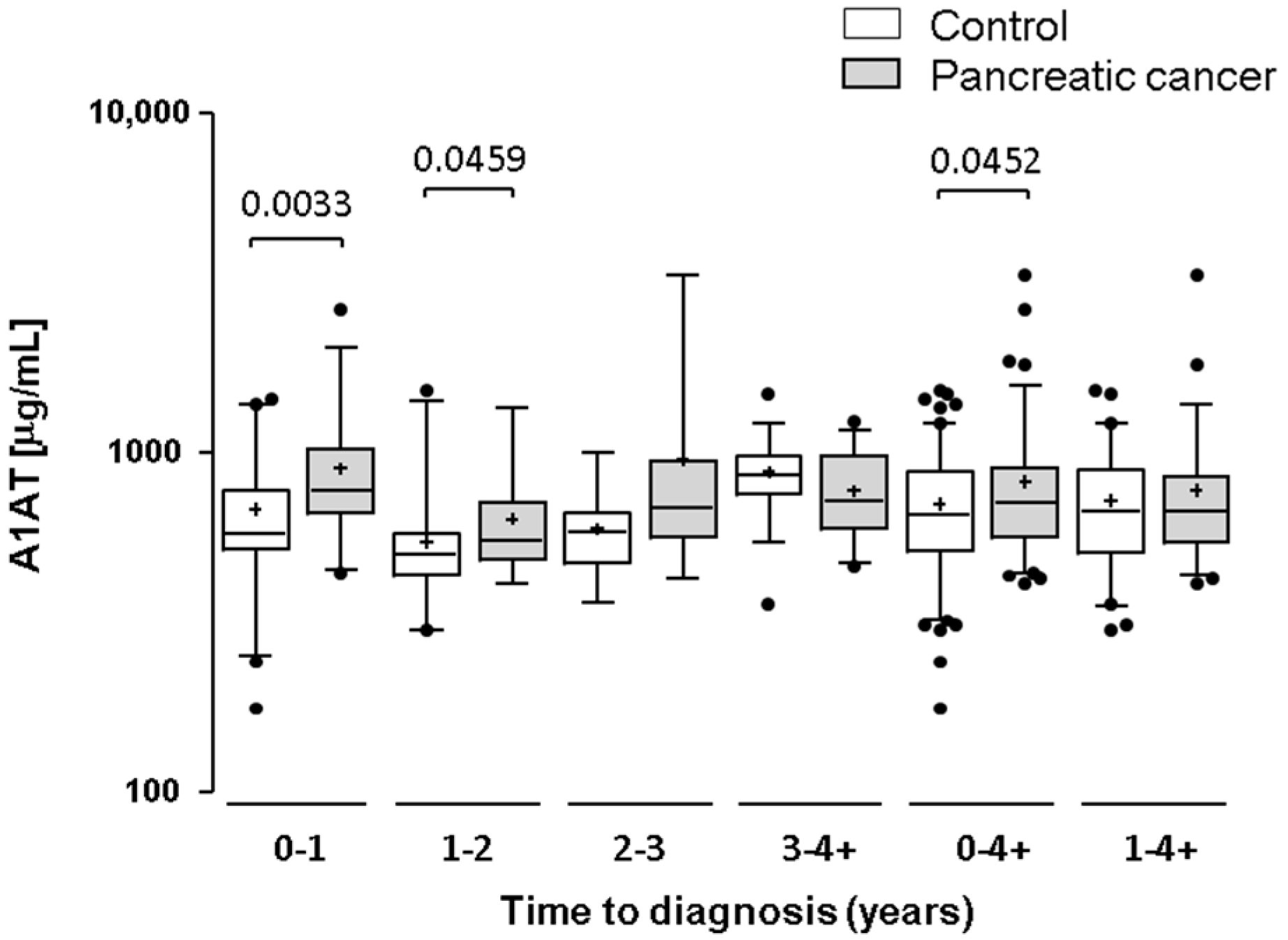
2.3. Testing α-1-Antitrypsin as an Early Biomarker of Pancreatic Cancer
3. Discussion
4. Materials and Methods
4.1. Ethics and Sample Set
4.2. Serum Immunodepletion
4.3. Digestion, Tandem Mass Tag Labelling, and Sample Clean-Up
4.4. Titanium Oxide (TiO2) Enrichment
4.5. Strong Cation Exchange (SCX) and High pH RP-LC Fractionation
4.6. LC-MS/MS Analysis
4.7. Mass spectrometry Data Analysis
4.8. Serum Assays and Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AUC | area under the ROC curve |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| PDAC | pancreatic ductal adenocarcinoma |
| ROC | receiver operating characteristics |
| SCX | strong cation exchange |
| TEAB | triethylammonium bicarbonate |
| TCEP | tris(2-carboxyethyl)phosphine |
| TiO2 | titanium oxide |
| TMT | tandem mass tags |
| TFA | trifluoroacetic acid |
| UKCTOCS | UK Collaborative Trial of Ovarian Cancer Screening |
References
- CRUK. Pancreatic Cancer Statistics. Available online: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer (accessed on 13 November 2017).
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Baeckstrom, D.; Hansson, G.C.; Nilsson, O.; Johansson, C.; Gendler, S.J.; Lindholm, L. Purification and characterization of a membrane-bound and a secreted mucin-type glycoprotein carrying the carcinoma-associated sialyl-Lea epitope on distinct core proteins. J. Biol. Chem. 1991, 266, 21537–21547. [Google Scholar] [PubMed]
- Baeckstrom, D.; Nilsson, O.; Price, M.R.; Lindholm, L.; Hansson, G.C. Discrimination of MUC1 mucins from other sialyl-Lea-carrying glycoproteins produced by colon carcinoma cells using a novel monoclonal antibody. Cancer Res. 1993, 53, 755–761. [Google Scholar] [PubMed]
- Ho, J.J.; Siddiki, B.; Kim, Y.S. Association of sialyl-Lewisa and sialyl-Lewisx with MUC-1 apomucin ina pancreatic cancer cell line. Cancer Res. 1995, 55, 3659–3663. [Google Scholar] [PubMed]
- Ballehaninna, U.K.; Chamberlain, R.S. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. J. Gastrointest. Oncol. 2012, 3, 105–119. [Google Scholar] [PubMed]
- Duffy, M.J. Carcinoembryonic antigen as a marker for colorectal cancer: Is it clinically useful? Clin. Chem. 2001, 47, 624–630. [Google Scholar] [PubMed]
- Lamerz, R. Role of tumour markers, cytogenetics. Ann. Oncol. 1999, 10, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, W. The clinical utility of the CA 19-9 tumor-associated antigen. Am. J. Gastroenterol. 1990, 85, 350–355. [Google Scholar] [PubMed]
- DiMagno, E.P.; Reber, H.A.; Tempero, M.A. AGA technical review on the epidemiology, diagnosis, and treatment of pancreatic ductal adenocarcinoma. American gastroenterological association. Gastroenterology 1999, 117, 1464–1484. [Google Scholar] [PubMed]
- Chang, C.Y.; Huang, S.P.; Chiu, H.M.; Lee, Y.C.; Chen, M.F.; Lin, J.T. Low efficacy of serum levels of CA 19-9 in prediction of malignant diseases in asymptomatic population in taiwan. Hepatogastroenterology 2006, 53, 1–4. [Google Scholar] [PubMed]
- Kim, J.E.; Lee, K.T.; Lee, J.K.; Paik, S.W.; Rhee, J.C.; Choi, K.W. Clinical usefulness of carbohydrate antigen 19-9 as a screening test for pancreatic cancer in an asymptomatic population. J. Gastroenterol. Hepatol. 2004, 19, 182–186. [Google Scholar] [CrossRef]
- Satake, K.; Takeuchi, T.; Homma, T.; Ozaki, H. CA19-9 as a screening and diagnostic tool in symptomatic patients: The Japanese experience. Pancreas 1994, 9, 703–706. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, D.P.; Sandanayake, N.S.; Jenkinson, C.; Gentry-Maharaj, A.; Apostolidou, S.; Fourkala, E.O.; Camuzeaux, S.; Blyuss, O.; Gunu, R.; Dawnay, A.; et al. Serum CA19-9 is significantly up-regulated up to 2 years prior to diagnosis with pancreatic cancer: Implications for early disease detection. Clin. Cancer Res. 2015, 21, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Cwik, G.; Wallner, G.; Skoczylas, T.; Ciechanski, A.; Zinkiewicz, K. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch. Surg. 2006, 141, 968–973. [Google Scholar] [CrossRef]
- Haga, Y.; Sakamoto, K.; Egami, H.; Yoshimura, R.; Mori, K.; Akagi, M. Clinical significance of serum CA125 values in patients with cancers of the digestive system. Am. J. Med. Sci. 1986, 292, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Haglund, C. Tumour marker antigen CA125 in pancreatic cancer: A comparison with CA19-9 and CEA. Br. J. Cancer 1986, 54, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Haga, Y.; Yoshimura, R.; Egami, H.; Yokoyama, Y.; Akagi, M. Comparative effectiveness of the tumour diagnostics, CA 19-9, CA 125 and carcinoembryonic antigen in patients with diseases of the digestive system. Gut 1987, 28, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Yasue, M.; Sakamoto, J.; Teramukai, S.; Morimoto, T.; Yasui, K.; Kuno, N.; Kurimoto, K.; Ohashi, Y. Prognostic values of preoperative and postoperative CEA and CA19.9 levels in pancreatic cancer. Pancreas 1994, 9, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Simeone, D.M.; Ji, B.; Banerjee, M.; Arumugam, T.; Li, D.; Anderson, M.A.; Bamberger, A.M.; Greenson, J.; Brand, R.E.; Ramachandran, V.; et al. CEACAM1, a novel serum biomarker for pancreatic cancer. Pancreas 2007, 34, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.V.; Modrak, D.E.; Ying, Z.; Cardillo, T.M.; Sharkey, R.M.; Goldenberg, D.M. New MUC1 serum immunoassay differentiates pancreatic cancer from pancreatitis. J. Clin. Oncol. 2006, 24, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Koopmann, J.; Fedarko, N.S.; Jain, A.; Maitra, A.; Iacobuzio-Donahue, C.; Rahman, A.; Hruban, R.H.; Yeo, C.J.; Goggins, M. Evaluation of osteopontin as biomarker for pancreatic adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2004, 13, 487–491. [Google Scholar] [CrossRef]
- Koopmann, J.; Buckhaults, P.; Brown, D.A.; Zahurak, M.L.; Sato, N.; Fukushima, N.; Sokoll, L.J.; Chan, D.W.; Yeo, C.J.; Hruban, R.H.; et al. Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin. Cancer Res. 2004, 10, 2386–2392. [Google Scholar] [CrossRef]
- Faca, V.M.; Song, K.S.; Wang, H.; Zhang, Q.; Krasnoselsky, A.L.; Newcomb, L.F.; Plentz, R.R.; Gurumurthy, S.; Redston, M.S.; Pitteri, S.J.; et al. A mouse to human search for plasma proteome changes associated with pancreatic tumor development. PLoS Med. 2008, 5, e123. [Google Scholar]
- Rosty, C.; Christa, L.; Kuzdzal, S.; Baldwin, W.M.; Zahurak, M.L.; Carnot, F.; Chan, D.W.; Canto, M.; Lillemoe, K.D.; Cameron, J.L.; et al. Identification of hepatocarcinoma intestine pancreas/pancreatitis associated protein 1 as a biomarker for pancreatic ductal adenocarcinoma by protein biochip technology. Cancer Res. 2002, 62, 1868–1875. [Google Scholar]
- Cerwenka, H.; Aigner, R.; Bacher, H.; Werkgartner, G.; el-Shabrawi, A.; Quehenberger, F.; Mischinger, H.J. TUM2-PK (pyruvate kinase type tumor M2), CA19-9 and CEA in patients with benign, malignant and metastasizing pancreatic lesions. Anticancer Res. 1999, 19, 849–851. [Google Scholar]
- Rudd, P.M.; Dwek, R.A. Glycosylation: Heterogeneity and the 3D structure of proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 1–100. [Google Scholar]
- Zhao, J.; Qiu, W.; Simeone, D.M.; Lubman, D.M. N-linked glycosylation profiling of pancreatic cancer serum using capillary liquid phase separation coupled with mass spectrometric analysis. J. Proteome Res. 2007, 6, 1126–1138. [Google Scholar] [CrossRef]
- Sarrats, A.; Saldova, R.; Pla, E.; Fort, E.; Harvey, D.J.; Struwe, W.B.; de Llorens, R.; Rudd, P.M.; Peracaula, R. Glycosylation of liver acute-phase proteins in pancreatic cancer and chronic pancreatitis. Proteom. Clin. Appl. 2010, 4, 432–448. [Google Scholar] [CrossRef]
- Terao, N.; Takamatsu, S.; Minehira, T.; Sobajima, T.; Nakayama, K.; Kamada, Y.; Miyoshi, E. Fucosylation is a common glycosylation type in pancreatic cancer stem cell-like phenotypes. World J. Gastroenterol. 2015, 21, 3876–3887. [Google Scholar] [CrossRef]
- Pan, S.; Brentnall, T.A.; Chen, R. Glycoproteins and glycoproteomics in pancreatic cancer. World J. Gastroenterol. 2016, 22, 9288–9299. [Google Scholar] [CrossRef]
- Pan, S.; Chen, R.; Tamura, Y.; Crispin, D.A.; Lai, L.A.; May, D.H.; McIntosh, M.W.; Goodlett, D.R.; Brentnall, T.A. Quantitative glycoproteomics analysis reveals changes in N-glycosylation level associated with pancreatic ductal adenocarcinoma. J. Proteome Res. 2014, 13, 1293–1306. [Google Scholar] [CrossRef]
- Lau, K.S.; Dennis, J.W. N-glycans in cancer progression. Glycobiology 2008, 18, 750–760. [Google Scholar] [CrossRef]
- Dimitroff, C.J. Galectin-binding O-glycosylations as regulators of malignancy. Cancer Res. 2015, 75, 3195–3202. [Google Scholar] [CrossRef]
- Chia, J.; Goh, G.; Bard, F. Short O-galnac glycans: Regulation and role in tumor development and clinical perspectives. Biochim. Biophys. Acta 2016, 1860, 1623–1639. [Google Scholar] [CrossRef]
- Fry, S.A.; Afrough, B.; Lomax-Browne, H.J.; Timms, J.F.; Velentzis, L.S.; Leathem, A.J. Lectin microarray profiling of metastatic breast cancers. Glycobiology 2011, 21, 1060–1070. [Google Scholar] [CrossRef]
- Drake, R.R.; Schwegler, E.E.; Malik, G.; Diaz, J.; Block, T.; Mehta, A.; Semmes, O.J. Lectin capture strategies combined with mass spectrometry for the discovery of serum glycoprotein biomarkers. Mol. Cell. Proteom. 2006, 5, 1957–1967. [Google Scholar] [CrossRef]
- Larsen, M.R.; Jensen, S.S.; Jakobsen, L.A.; Heegaard, N.H. Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol. Cell. Proteom. 2007, 6, 1778–1787. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.J.; Martin, D.B.; Aebersold, R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003, 21, 660–666. [Google Scholar] [CrossRef]
- Menon, U.; Gentry-Maharaj, A.; Hallett, R.; Ryan, A.; Burnell, M.; Sharma, A.; Lewis, S.; Davies, S.; Philpott, S.; Lopes, A.; et al. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancers: Results of the prevalence screen of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). Lancet Oncol. 2009, 10, 327–340. [Google Scholar] [CrossRef]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef]
- Palmisano, G.; Melo-Braga, M.N.; Engholm-Keller, K.; Parker, B.L.; Larsen, M.R. Chemical deamidation: A common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J. Proteome Res. 2012, 11, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Kontro, H.; Joenvaara, S.; Haglund, C.; Renkonen, R. Comparison of sialylated N-glycopeptide levels in serum of pancreatic cancer patients, acute pancreatitis patients, and healthy controls. Proteomics 2014, 14, 1713–1723. [Google Scholar]
- Chen, A.; Kozak, D.; Battersby, B.J.; Forrest, R.M.; Scholler, N.; Urban, N.; Trau, M. Antifouling surface layers for improved signal-to-noise of particle-based immunoassays. Langmuir 2009, 25, 13510–13515. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, Y.; Mashiko, T.; Danbara, M.; Takayama, Y.; Ohtani, S.; Egawa, S.; Baba, S.; Akahoshi, T. Changes in serum IgG oligosaccharide chains with prostate cancer progression. Anticancer Res. 2004, 24, 3135–3139. [Google Scholar] [PubMed]
- Kodar, K.; Izotova, J.; Klaamas, K.; Sergeyev, B.; Jarvekulg, L.; Kurtenkov, O. Aberrant glycosylation of the anti-Thomsen-Friedenreich glycotope immunoglobulin G in gastric cancer patients. World J. Gastroenterol. 2013, 19, 3573–3582. [Google Scholar] [PubMed]
- Bones, J.; Byrne, J.C.; O'Donoghue, N.; McManus, C.; Scaife, C.; Boissin, H.; Nastase, A.; Rudd, P.M. Glycomic and glycoproteomic analysis of serum from patients with stomach cancer reveals potential markers arising from host defense response mechanisms. J. Proteome Res. 2011, 10, 1246–1265. [Google Scholar] [PubMed]
- Saldova, R.; Royle, L.; Radcliffe, C.M.; Abd Hamid, U.M.; Evans, R.; Arnold, J.N.; Banks, R.E.; Hutson, R.; Harvey, D.J.; Antrobus, R.; et al. Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and igg. Glycobiology 2007, 17, 1344–1356. [Google Scholar] [PubMed]
- Arnold, J.N.; Dwek, R.A.; Rudd, P.M.; Sim, R.B. Mannan binding lectin and its interaction with immunoglobulins in health and in disease. Immunol. Lett. 2006, 106, 103–110. [Google Scholar] [PubMed]
- Sinclair, J.; Timms, J.F. Quantitative profiling of serum samples using tmt protein labelling, fractionation and LC-MS/MS. Methods 2011, 54, 361–369. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Protein Name | Unique Peptides | † PSMs | Score | Ratio (0–0.5 years vs. † Ctrl) | Ratio (0.5–1 years vs. Ctrl) | Ratio (1–2 years vs. Ctrl) | Ratio (2–3 years vs. Ctrl) | Ratio (3+ years vs. Ctrl) |
|---|---|---|---|---|---|---|---|---|---|
| P01009 | α-1-Antitrypsin | 25 | 1183 | 27.0 | 3.294 | 2.207 | 2.000 | 2.230 | 1.930 |
| P00738 | Haptoglobin | 10 | 951 | 25.8 | 2.720 | 2.077 | 1.551 | 2.076 | 1.636 |
| P00739 | Haptoglobin-related protein | 4 | 354 | 22.8 | 2.832 | 2.053 | 1.592 | 1.959 | 1.660 |
| P02763 | α-1-Acid glycoprotein 1 | 7 | 239 | 19.0 | 1.909 | 1.554 | 1.294 | 1.662 | 1.319 |
| P01857 | Igγ-1 chain C region | 7 | 738 | 18.8 | 1.714 | 3.338 | 1.613 | 1.202 | 1.535 |
| P01860 | Igγ-3 chain C region | 37 | 385 | 18.6 | 1.726 | 3.315 | 1.653 | 1.264 | 1.538 |
| P01871 | Igμ chain C region | 18 | 139 | 18.4 | 1.558 | 1.637 | 1.306 | 1.304 | 1.273 |
| P02787 | Serotransferrin | 53 | 1610 | 17.8 | 1.723 | 2.017 | 1.466 | 1.200 | 1.416 |
| P19652 | α-1-acid glycoprotein 2 | 20 | 196 | 17.8 | 1.819 | 1.552 | 1.326 | 1.608 | 1.376 |
| P01023 | α-2-macroglobulin | 5 | 142 | 17.8 | 1.707 | 1.605 | 1.337 | 1.232 | 1.340 |
| P08603 | Complement factor H | 5 | 101 | 17.0 | 1.503 | 1.373 | 1.234 | 1.330 | 1.378 |
| P01880 | Igδ chain C region | 16 | 49 | 17.0 | 1.892 | 2.748 | 1.942 | 1.557 | 1.405 |
| P01861 | Igγ-4 chain C region | 32 | 471 | 17.0 | 1.558 | 2.878 | 1.595 | 1.246 | 1.478 |
| P00748 | Coagulation factor XII | 15 | 657 | 17.0 | 1.489 | 1.359 | 1.241 | 1.351 | 1.125 |
| P05543 | Thyroxine-binding globulin | 13 | 83 | 16.8 | 0.828 | 0.765 | 0.769 | 0.837 | 0.884 |
| P10643 | Complement component 7 | 16 | 146 | 16.8 | 1.432 | 1.341 | 1.508 | 1.350 | 1.260 |
| P43652 | Afamin | 31 | 676 | 16.4 | 0.811 | 0.790 | 0.822 | 0.910 | 0.998 |
| P02765 | α-2-HS-glycoprotein | 13 | 18 | 16.2 | 0.970 | 0.975 | 0.977 | 1.025 | 1.118 |
| P43251 | Biotinidase | 12 | 64 | 16.2 | 0.957 | 0.931 | 0.986 | 1.013 | 1.039 |
| P04114 | Apolipoprotein B100 | 20 | 73 | 16.2 | 1.152 | 1.333 | 1.510 | 1.498 | 1.146 |
| Sequence | Accession | Protein Name | † PSMs | Score | Ratio (0–0.5 years vs. Ctrl) | Ratio (0.5–1 years vs. Ctrl) | Ratio (1–2 years vs. Ctrl) | Ratio (2–3 years vs. Ctrl) | Ratio (3+ years vs. Ctrl) |
|---|---|---|---|---|---|---|---|---|---|
| yLGnATAIFFLPDEGk | P01009 | α-1-Antitrypsin | 27 | 31 | 4.06 | 2.69 | 2.39 | 2.63 | 2.53 |
| nLFLnHSEnATAk | P00738 | Haptoglobin | 42 | 28.8 | 3.28 | 2.38 | 1.67 | 2.36 | 1.79 |
| nLFLnHSEnATAk | P00738 | Haptoglobin | 43 | 24 | 2.66 | 1.90 | 1.58 | 1.91 | 1.55 |
| vVLHPnYSQVDIGLIk | P00738 | Haptoglobin | 16 | 20.2 | 2.60 | 1.84 | 1.31 | 1.92 | 1.44 |
| eEqFnSTFR | P01859 | Igγ-2 chain C region | 16 | 19.6 | 0.46 | 0.50 | 0.53 | 0.60 | 0.58 |
| lGAcnDTLQQLmEVFk | P01008 | Antithrombin-III | 7 | 19 | 0.81 | 0.70 | 0.71 | 0.73 | 0.75 |
| eTFFnLSk | Q9UK55 | Protein Z-dependent protease inhibitor | 3 | 19 | 1.91 | 2.05 | 2.53 | 2.19 | 1.98 |
| enGTISR | P02763 | α-1-Acid glycoprotein 1 | 57 | 19 | 2.25 | 1.74 | 1.48 | 1.99 | 1.45 |
| qDqcIYnTTYLNVQR | P02763 | α-1-Acid glycoprotein 1 | 149 | 18.6 | 1.98 | 1.61 | 1.36 | 1.72 | 1.33 |
| lGHcPDPVLVnGEFSSSGPVnVSDk | P20851 | C4b-binding protein β chain | 8 | 18.4 | 1.66 | 1.76 | 1.37 | 1.55 | 1.44 |
| qNQcFYnSSYLNVQR | P19652 | α-1-Acid glycoprotein 2 | 24 | 18 | 1.93 | 1.57 | 1.20 | 1.53 | 1.26 |
| aAIPSALDTnSSk | Q08380 | Galectin-3-binding protein | 24 | 17.6 | 0.90 | 0.94 | 0.99 | 1.00 | 1.09 |
| enGTVSR | P19652 | α-1-Acid glycoprotein 2 | 22 | 17.2 | 1.93 | 1.64 | 1.33 | 1.67 | 1.45 |
| eDAAVncTDISVQk | Q86VB7 | Scavenger receptor cysteine-rich type 1 | 3 | 17 | 0.61 | 0.63 | 0.64 | 0.65 | 0.72 |
| vcQDcPLLAPLnDTR | P02765 | α-2-HS-glycoprotein | 49 | 17 | 0.93 | 1.09 | 0.91 | 1.12 | 1.24 |
| dIEnFnSTQk | P43652 | Afamin | 56 | 17 | 0.80 | 0.74 | 0.83 | 0.87 | 0.94 |
| nYTLTGR | P10643 | Complement component C7 | 3 | 17 | 2.15 | 1.94 | 2.07 | 1.75 | 1.55 |
| aDGTVNQIEGEATPVnLTEPAk | P05090 | Apolipoprotein D | 66 | 16.8 | 1.24 | 1.15 | 1.12 | 1.00 | 1.18 |
| enLTAPGSDSAVFFEQGTTR | P00450 | Ceruloplasmin | 131 | 16.8 | 0.88 | 0.85 | 0.85 | 0.89 | 0.98 |
| nVTAEQAR | P00748 | Coagulation factor XII | 10 | 16.8 | 1.67 | 1.64 | 1.31 | 1.49 | 1.16 |
| Time Group | No. Case Samples | Marker Model | AUC | Sensitivity at 0.95 Specificity |
|---|---|---|---|---|
| 0–1 years | 34 | CA9-9 | 0.799 | 0.647 |
| CA19-9, A1AT | 0.88 | 0.647 | ||
| 1–2 years | 17 | CA9-9 | 0.579 | 0.176 |
| CA19-9, A1AT | 0.664 | 0.235 | ||
| 2+ years | 41 | CA9-9 | 0.618 | 0.073 |
| CA19-9, A1AT | 0.644 | 0.146 | ||
| 0–2 years | 51 | CA9-9 | 0.726 | 0.49 |
| CA19-9, A1AT | 0.76 | 0.471 | ||
| 0–3 years | 64 | CA9-9 | 0.7 | 0.422 |
| CA19-9, A1AT | 0.738 | 0.438 | ||
| 0–4+ years (All) | 92 | CA9-9 | 0.678 | 0.304 |
| CA19-9, A1AT | 0.709 | 0.315 |
| Variable | Cases | Controls | p Value |
|---|---|---|---|
| No. individuals | 154 | 304 | - |
| No. samples | 261 | 304 | - |
| Tumour site | |||
| Tail | 8 | na | - |
| Body | 10 | na | - |
| Head | 65 | na | - |
| Unspecified | 71 | na | - |
| Mean time to spin (h) (range) | 21.8 (0.5–47) | 22.0 (6.9–47) | 0.62 |
| Mean age at sample draw (years) (range) | 64.64 (51.6–74.4) | 62.67 (50.6–77.5) | 0.049 |
| Mean BMI (kg/m2) (range) | 27.1 (18.6–42.7) | 26.7 (17.9–44.4) | 0.446 |
| Mean time from sample collection to diagnosis (months) (range) | 25.5 (0–79) | na | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, S.; Whitwell, H.J.; Cuenco, J.; Gentry-Maharaj, A.; Menon, U.; Pereira, S.P.; Gaspari, M.; Timms, J.F. Evidence of Altered Glycosylation of Serum Proteins Prior to Pancreatic Cancer Diagnosis. Int. J. Mol. Sci. 2017, 18, 2670. https://doi.org/10.3390/ijms18122670
Krishnan S, Whitwell HJ, Cuenco J, Gentry-Maharaj A, Menon U, Pereira SP, Gaspari M, Timms JF. Evidence of Altered Glycosylation of Serum Proteins Prior to Pancreatic Cancer Diagnosis. International Journal of Molecular Sciences. 2017; 18(12):2670. https://doi.org/10.3390/ijms18122670
Chicago/Turabian StyleKrishnan, Shibu, Harry J. Whitwell, Joy Cuenco, Aleksandra Gentry-Maharaj, Usha Menon, Stephen P. Pereira, Marco Gaspari, and John F. Timms. 2017. "Evidence of Altered Glycosylation of Serum Proteins Prior to Pancreatic Cancer Diagnosis" International Journal of Molecular Sciences 18, no. 12: 2670. https://doi.org/10.3390/ijms18122670
APA StyleKrishnan, S., Whitwell, H. J., Cuenco, J., Gentry-Maharaj, A., Menon, U., Pereira, S. P., Gaspari, M., & Timms, J. F. (2017). Evidence of Altered Glycosylation of Serum Proteins Prior to Pancreatic Cancer Diagnosis. International Journal of Molecular Sciences, 18(12), 2670. https://doi.org/10.3390/ijms18122670