High-Throughput RNA-Seq Data Analysis of the Single Nucleotide Polymorphisms (SNPs) and Zygomorphic Flower Development in Pea (Pisum sativum L.)

Abstract
:
1. Introduction
2. Results
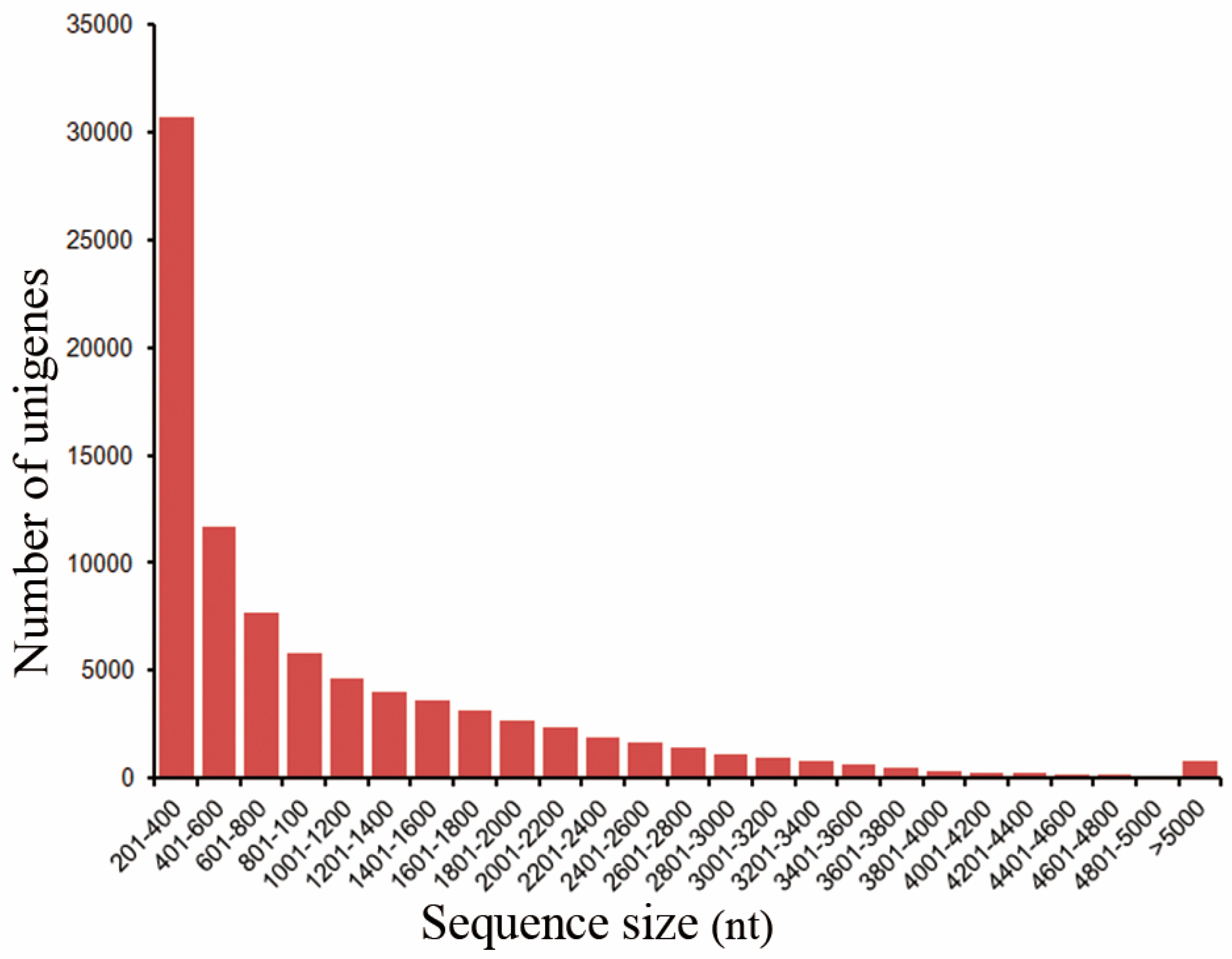
2.1. De Novo Assembly of Illumina Paired-End Reads and Unigene Annotation
2.2. SNP Identification
2.3. Comparative Genome Analysis between Pea and Medicago
2.4. Identification of DEGs in the Three Types of Pea Petals
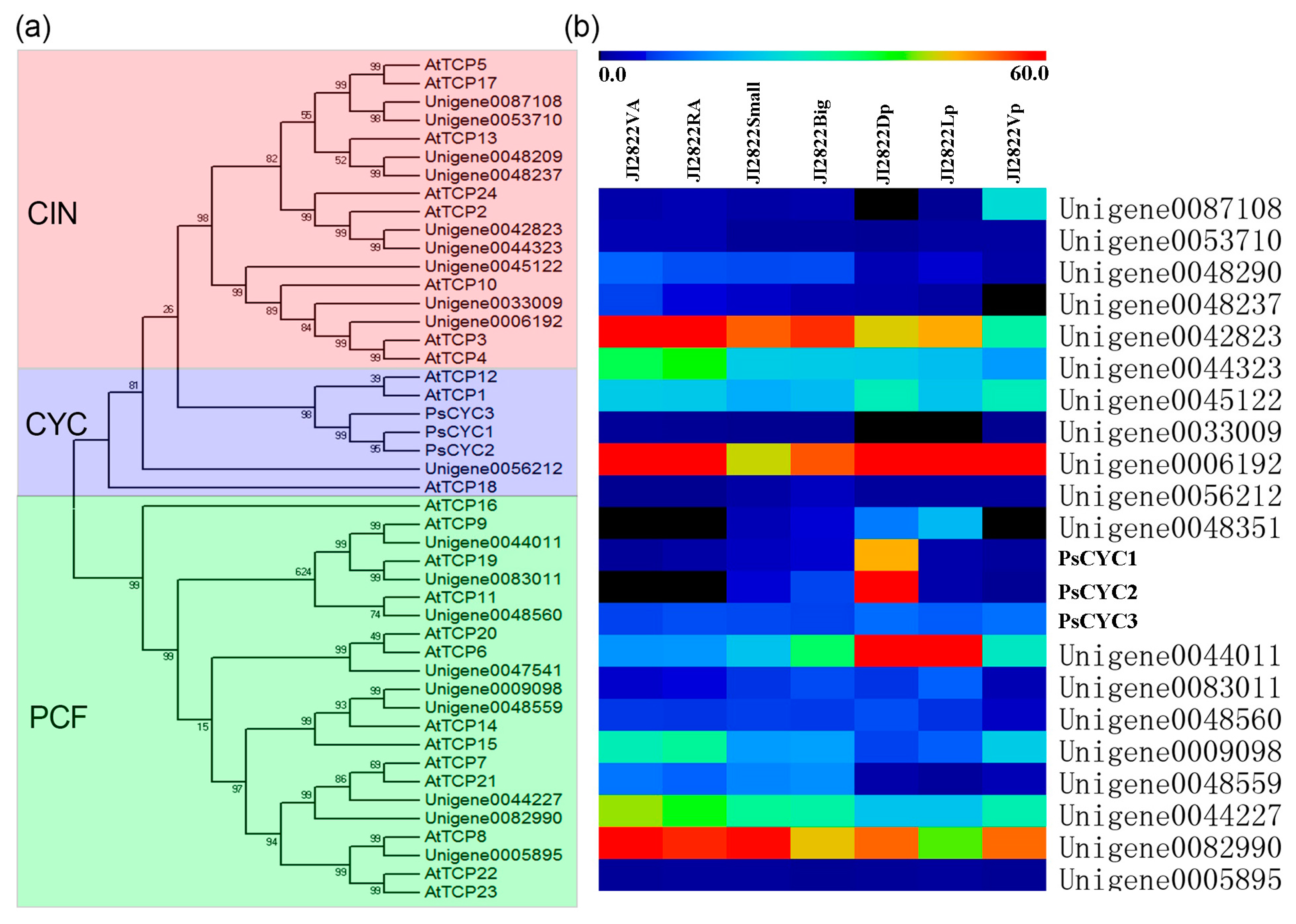
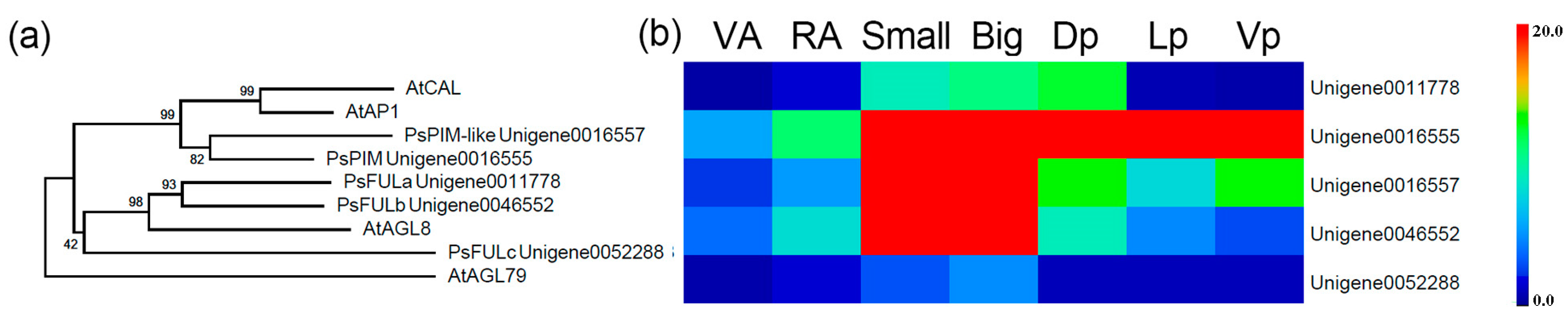
2.5. Determination of Gene Categories That Were Differentially Expressed in the Petals
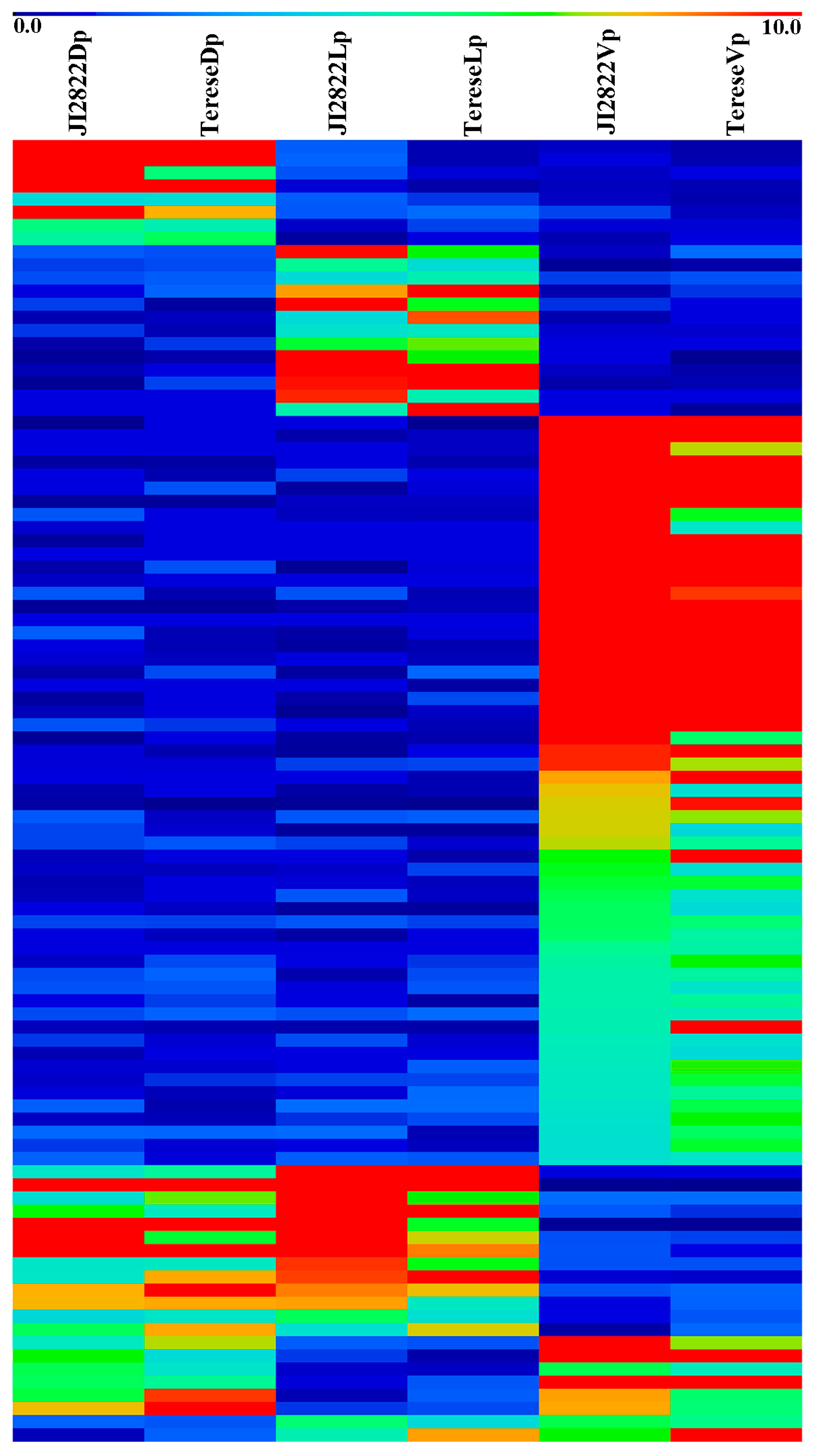
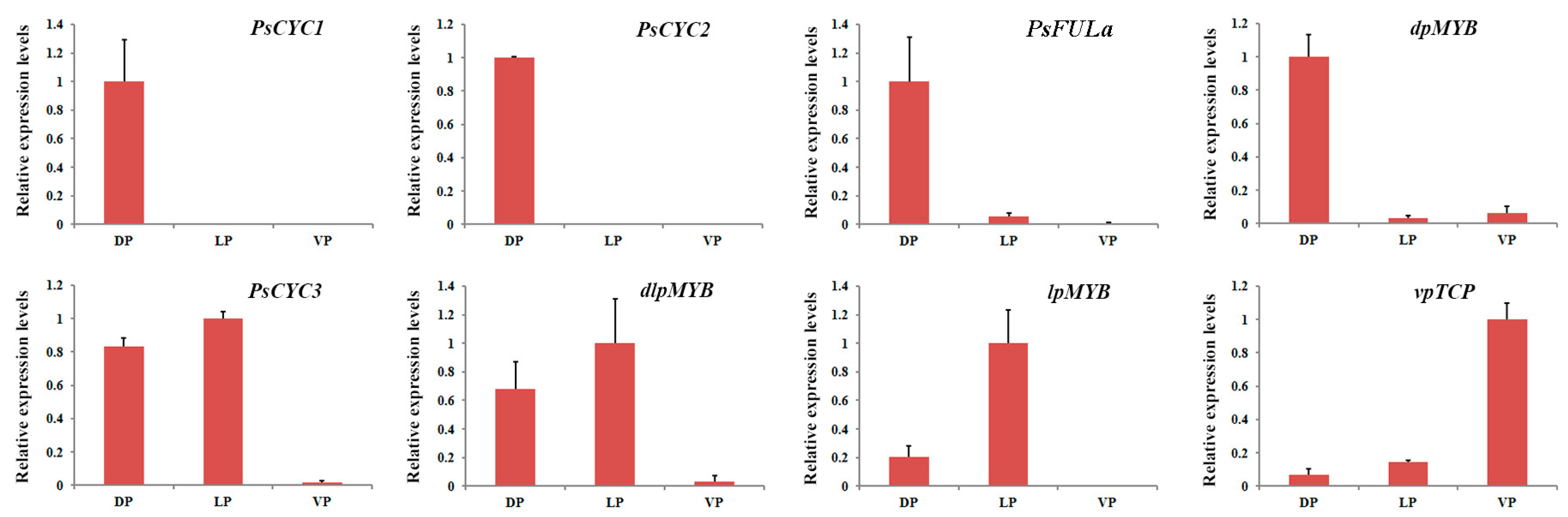
2.6. Expression Analysis of Key DEGs in Petals via qRT-PCR
3. Discussion
3.1. SNP Variations in Pea and Comparative Genome Analysis
3.2. Floral Zygomorphy as Revealed by Transcriptional Profiling in Pea
4. Materials and Methods
4.1. Plant Materials and RNA Extraction
4.2. Illumina Sequencing and De Novo Assembly
4.3. SNP Discovery
4.4. Comparative Genome Analysis and the Pea Consensus Map
4.5. Functional Annotation
4.6. Phylogeny Analysis
4.7. Quantitative RT-PCR
4.8. Differentially Expressed Gene (DEG) Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Z.; Luo, Y.; Li, X.; Wang, L.; Xu, S.; Yang, J.; Weng, L.; Sato, S.; Tabata, S.; Ambrose, M.; et al. Genetic control of floral zygomorphy in pea (Pisum sativum L.). Proc. Natl. Acad. Sci. USA 2008, 105, 10414–10419. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhuang, L.L.; Ambrose, M.; Rameau, C.; Hu, X.H.; Yang, J.; Luo, D. Genetic analysis of ELE mutants and comparative mapping of ele1 locus in the control of organ internal asymmetry in garden pea. J. Integr. Plant Biol. 2010, 52, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Domoney, C.; Knox, M.; Moreau, C.; Ambrose, M.; Palmer, S.; Smith, P.; Christodoulou, V.; Isaac, P.G.; Hegarty, M.; Blackmore, T.; et al. Exploiting a fast neutron mutant genetic resource in Pisum sativum (pea) for functional genomics. Funct. Plant Biol. 2013, 40, 1261–1270. [Google Scholar] [CrossRef]
- Constantin, G.D.; Krath, B.N.; MacFarlane, S.A.; Nicolaisen, M.; Johansen, I.E.; Lund, O.S. Virus-induced gene silencing as a tool for functional genomics in a legume species. Plant J. 2004, 4, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Meziadi, C.; Blanchet, S.; Richard, M.M.; Pilet-Nayel, M.L.; Geffroy, V.; Pflieger, S. Bean pod mottle virus: A new powerful tool for functional genomics studies in Pisum sativum. Plant Biotechnol. J. 2016, 14, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the paleopolyploid soybean. Nature 2010, 46, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Udvardi, M. Translating Medicago truncatula genomics to crop legumes. Curr. Opin. Plant Biol. 2009, 12, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.K.; Mun, J.H.; Kim, D.J.; Zhu, H.; Baek, J.M.; Mudge, J. Estimating genome conservation between crop and model legume species. Proc. Natl. Acad. Sci. USA 2004, 1, 15289–15294. [Google Scholar] [CrossRef] [PubMed]
- Kaló, P.; Seres, A.; Taylor, S.A.; Jakab, J.; Kevei, Z.; Kereszt, A.; Endre, G.; Ellis, T.H.; Kiss, G.B. Comparative mapping between Medicago sativum and Pisum sativum. Mol. Genet. Genom. 2004, 272, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef] [PubMed]
- Cronk, Q.; Ojeda, I.; Pennington, R.T. Legume comparative genomics: Progress in phylogenetics and phylogenomics. Curr. Opin. Plant Biol. 2006, 9, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Close, T.J.; Singh, N.K.; Hoisington, D.A.; Cook, D.R. Orphan legume crops enter the genomics era! Curr. Opin. Plant Biol. 2009, 12, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.L.; Ambrose, M.; Rameau, C.; Weng, L.; Yang, J.; Hu, X.H.; Luo, D.; Li, X. LATHYROIDES, encoding a WUSCHEL-related Homeobox1 transcription factor, controls organ lateral growth, and regulates tendril and dorsal petal identities in garden pea (Pisum sativum L.). Mol. Plant 2012, 5, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Bordat, A.; Savois, V.; Nicolas, M.; Salse, J.; Chauveau, A.; Bourgeois, M.; Potier, J.; Houtin, H.; Rond, C.; Murat, F. Translational genomics in Legumes allowed placing in silico 5460 unigenes on the Pea functional map and identified candidate genes in Pisum sativum L. G3 (Bethesda) 2011, 1, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Leonforte, A.; Sudheesh, S.; Cogan, N.O.; Salisbury, P.A.; Nicolas, M.E.; Materne, M.; Forster, J.W.; Kaur, S. SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (Pisum sativum L.). BMC Plant Biol. 2013, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.; Rivière, N.; Baranger, A.; Aubert, G.; Burstin, J.; Cornet, L.; Lavaud, C.; Lejeune-Hénaut, I.; Martinant, J.P.; Pichon, J.P.; et al. Transcriptome sequencing for high throughput SNP development and genetic mapping in Pea. BMC Genom. 2014, 15, 126. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, A.; Ramsay, L.; Sanderson, L.A.; Stonehouse, R.; Li, R.; Condie, J.; Shunmugam, A.S.; Liu, Y.; Jha, A.B.; Diapari, M.; et al. Gene-based SNP discovery and genetic mapping in pea. Theor. Appl. Genet. 2014, 127, 2225–2241. [Google Scholar] [CrossRef] [PubMed]
- Burstin, J.; Salloignon, P.; Chabert-Martinello, M.; Magnin-Robert, J.B.; Siol, M.; Jacquin, F.; Chauveau, A.; Pont, C.; Aubert, G.; Delaitre, C.; et al. Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genom. 2015, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Sudheesh, S.; Sawbridge, T.I.; Cogan, N.O.; Kennedy, P.; Forster, J.W.; Kaur, S. De novo assembly and characterization of the field pea transcriptome using RNA-Seq. BMC Genom. 2015, 16, 611. [Google Scholar] [CrossRef] [PubMed]
- Boutet, G.; Alves Carvalho, S.; Falque, M.; Peterlongo, P.; Lhuillier, E.; Bouchez, O.; Lavaud, C.; Pilet-Nayel, M.L.; Rivière, N.; Baranger, A. SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population. BMC Genom. 2016, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, S.C.; Gaiti, F.; Beveridge, C.A.; Tanurdzic, M. De novo transcriptome assembly reveals high transcriptional complexity in Pisum sativum axillary buds and shows rapid changes in expression of diurnally regulated genes. BMC Genom. 2017, 18, 221. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.C. Floral development in legumes. Plant Physiol. 2003, 131, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Carpenter, R.; Vincent, C.; Copsey, L.; Coen, E. Origin of floral asymmetry in Antirrhinum. Nature 1996, 383, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Carpenter, R.; Copsey, L.; Vincent, C.; Clark, J.; Coen, E. Control of organ asymmetry in flowers of Antirrhinum. Cell 1999, 99, 367–376. [Google Scholar] [CrossRef]
- Howarth, D.G.; Donoghue, M.J. Phylogenetic analysis of the ‘ECE’ (CYC/TB1) clade reveals duplications predating the core eudicots. Proc. Natl. Acad. Sci. USA 2006, 103, 9101–9106. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Zachgo, S. Control of corolla monosymmetry in the Brassicaceae Iberis amara. Proc. Natl. Acad. Sci. USA 2007, 104, 16714–16719. [Google Scholar] [CrossRef] [PubMed]
- Broholm, S.K.; Tähtiharju, S.; Laitinen, R.A.; Albert, V.A.; Teeri, T.H.; Elomaa, P. A TCP transcription factor controls flowers type specification along the radial axis of the Gerbera (Asteraceae) inflorescence. Proc. Natl. Acad. Sci. USA 2008, 10, 9117–9122. [Google Scholar] [CrossRef] [PubMed]
- Presto, J.C.; Hileman, L.C. Developmental genetics of floral symmetry evolution. Trends Plant Sci. 2009, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Citerne, H.L.; Pennington, R.T.; Cronk, Q.C. An apparent reversal in floral symmetry in the legume Cadia is a homeotic transformation. Proc. Natl. Acad. Sci. USA 2006, 103, 12017–12020. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhao, Z.; Tian, Z.; Xu, S.; Luo, Y.; Cai, Z.; Wang, Y.; Yang, J.; Wang, Z.; Weng, L.; et al. Control of petal shape and floral zygomorphy in Lotus japonicus. Proc. Natl. Acad. Sci. USA 2006, 103, 4970–4975. [Google Scholar] [CrossRef] [PubMed]
- Loridon, K.; McPhee, K.; Morin, J.; Dubreuil, P.; Pilet-Nayel, M.L.; Aubert, G.; Rameau, C.; Baranger, A.; Coyne, C.; Lejeune-Hènaut, I.; et al. Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Theor. Appl. Genet. 2005, 111, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Martin-Trillo, M.; Cubas, P. TCP genes: A family snapshot ten years later. Trends Plant Sci. 2010, 15, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, X.G.; Li, C.Q.; Liu, J.; Qiu, Z.J.; Dong, Y.; Wang, Y.Z. Distinct regulatory changes underlying differential expression of teosinte branched1-cycloidea-proliferating cell factor genes associated with petal variations in zygomorphic flowers of Petrocosmea spp. of the family gesneriaceae. Plant Physiol. 2015, 169, 2138–2151. [Google Scholar] [PubMed]
- Hecht, V.; Foucher, F.; Ferrándiz, C.; Macknight, R.; Navarro, C.; Morin, J.; Vardy, M.E.; Ellis, N.; Beltrán, J.P.; Rameau, C.; et al. Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol. 2005, 137, 1420–1434. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Geisler, M.; Springer, P.S. LATERAL ORGAN FUSION1 and LATERAL ORGAN FUSION2 function in lateral organ separation and axillary meristem formation in Arabidopsis. Development 2009, 136, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Galego, L.; Almeida, J. Role of DIVARICATA in the control of dorsoventral asymmetry in Antirrhinum flowers. Genes Dev. 2002, 16, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Corley, S.B.; Carpenter, R.; Copsey, L.; Coen, E. Floral asymmetry involves an interplay between TCP and MYB transcription factors in Antirrhinum. Proc. Natl. Acad. Sci. USA 2005, 102, 5068–5073. [Google Scholar] [CrossRef] [PubMed]
- Deulvot, C.; Charrel, H.; Marty, A.; Jacquin, F.; Donnadieu, C.; Lejeune-Hénaut, I.; Burstin, J.; Aubert, G. Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genom. 2010, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Aubert, G.; Morin, J.; Jacque, F.; Loridon, K.; Quillet, M.C.; Petit, A.; Rameau, C.; Lejeune-Hénaut, I.; Huguet, T.; Burstin, J. Functional mapping in pea, as an aid to the candidate gene selection and for investigating synteny with the model legume Medicago truncatula. Theor. Appl. Genet. 2006, 13, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Gao, S.; Xue, D.W.; Luo, D.; Li, L.T.; Ding, S.Y.; Yao, X.; Wilson, Z.A.; Qian, Q.; Zhang, D.B. RETARDED PALEA1 controls palea development and floral zygomorphy in rice. Plant Physiol. 2009, 149, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Kao, N.H.; Li, J.Y.; Hsu, W.H.; Liang, Y.L.; Wu, J.W. Characterization of the possible roles for B class MADS box genes in regulation of perianth formation in orchid. Plant Physiol. 2010, 152, 837–853. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.F.; Hsu, W.H.; Lee, Y.I.; Mao, W.T.; Yang, J.Y.; Li, J.Y.; Yang, C.H. Model for perianth formation in orchids. Nat. Plant 2015, 1, 15046. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Liu, K.W.; Li, Z.; Lohaus, R.; Hsiao, Y.Y.; Niu, S.C.; Wang, J.Y.; Lin, Y.C.; Xu, Q.; Chen, L.J.; et al. The Apostasia genome and the evolution of orchids. Nature 2017, 549, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.I.; Coen, E.S. The cycloidea gene can respond to a common dorsoventral prepattern in Antirrhinum. Plant J. 2002, 30, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Glover, B.J.; Linstead, P.; Martin, C. Flower colour intensity depends on specialized cell shape controlled by a Myb related transcription factor. Nature 1994, 369, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Glover, B.J.; Perez-Rodriguez, M.; Martin, C. Development of several epidermal cell types can be specified by the same MYB related plant transcription factor. Development 1998, 125, 3497–3508. [Google Scholar] [PubMed]
- Perez-Rodriguez, M.; Jaffe, F.W.; Butelli, E.; Glover, B.J.; Martin, C. Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development 2005, 132, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.; Perez-Rodriguez, M.; Bradley, D.; Venail, J.; Bailey, P.; Jin, H.L.; Koes, R.; Roberts, K.; Martin, C. Control of cell and petal morphogenesis by R2R3 MYB transcription factors. Development 2007, 134, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, I.; Francisco-Ortega, J.; Cronk, Q.C. Evolution of petal epidermal micromorphology in Leguminosae and its use as a marker of petal identity. Ann. Bot. 2009, 104, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.M.; Fox, S.; Hanna, A.I.; Baxter, C.; Coen, E. Evolution of regulatory interactions controlling floral asymmetry. Development 2005, 132, 5093–5101. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Number of Reads | Number of Reads Showing Unique Matches | Percentage |
|---|---|---|---|
| JI2822 Vegetative shoot apices (VA) | 32,214,992 | 22,922,794 | 71.16% |
| JI2822 Reproductive shoot apices (RA) | 20,576,714 | 15,127,796 | 73.52% |
| JI2822 2-mm floral buds (Small) | 29,167,060 | 20,502,301 | 70.29% |
| JI2822 5-mm floral buds (Big) | 33,818,056 | 24,224,028 | 71.63% |
| JI2822 Dorsal petals (Dp) | 31,476,682 | 22,061,407 | 70.09% |
| JI2822 Lateral petals (Lp) | 22,633,942 | 15,691,372 | 69.33% |
| JI2822 Ventral petals (Vp) | 23,089,582 | 16,568,145 | 71.76% |
| Terese Vegetative shoot apices (VA) | 26,326,994 | 17,622,043 | 66.94% |
| Terese Reproductive shoot apices (RA) | 38,704,866 | 26,011,448 | 67.20% |
| Terese 2-mm floral buds (Small) | 41,799,160 | 28,426,856 | 68.01% |
| Terese 5-mm floral buds (Big) | 52,957,978 | 36,260,749 | 68.47% |
| Terese Dorsal petals (Dp) | 21,246,110 | 14,305,314 | 67.33% |
| Terese Lateral petals (Lp) | 24,931,766 | 16,559,528 | 66.42% |
| Terese Ventral petals (Vp) | 24,393,024 | 16,574,182 | 67.95% |
| JI992 Vegetative shoot apices (VA) | 42,342,066 | 28,572,173 | 67.48% |
| JI992 Reproductive shoot apices (RA) | 56,082,530 | 24,545,471 | 43.77% |
| JI992 2-mm floral buds (Small) | 47,278,346 | 32,007,646 | 67.70% |
| JI992 5-mm floral buds (Big) | 48,317,046 | 33,808,043 | 69.97% |
| Total | 617,356,914 | 411,791,296 | 66.70% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, K.; Li, X.; Guo, W.; Su, S.; Luo, D. High-Throughput RNA-Seq Data Analysis of the Single Nucleotide Polymorphisms (SNPs) and Zygomorphic Flower Development in Pea (Pisum sativum L.). Int. J. Mol. Sci. 2017, 18, 2710. https://doi.org/10.3390/ijms18122710
Jiao K, Li X, Guo W, Su S, Luo D. High-Throughput RNA-Seq Data Analysis of the Single Nucleotide Polymorphisms (SNPs) and Zygomorphic Flower Development in Pea (Pisum sativum L.). International Journal of Molecular Sciences. 2017; 18(12):2710. https://doi.org/10.3390/ijms18122710
Chicago/Turabian StyleJiao, Keyuan, Xin Li, Wuxiu Guo, Shihao Su, and Da Luo. 2017. "High-Throughput RNA-Seq Data Analysis of the Single Nucleotide Polymorphisms (SNPs) and Zygomorphic Flower Development in Pea (Pisum sativum L.)" International Journal of Molecular Sciences 18, no. 12: 2710. https://doi.org/10.3390/ijms18122710
APA StyleJiao, K., Li, X., Guo, W., Su, S., & Luo, D. (2017). High-Throughput RNA-Seq Data Analysis of the Single Nucleotide Polymorphisms (SNPs) and Zygomorphic Flower Development in Pea (Pisum sativum L.). International Journal of Molecular Sciences, 18(12), 2710. https://doi.org/10.3390/ijms18122710