G Protein-Coupled Receptors at the Crossroad between Physiologic and Pathologic Angiogenesis: Old Paradigms and Emerging Concepts




Abstract
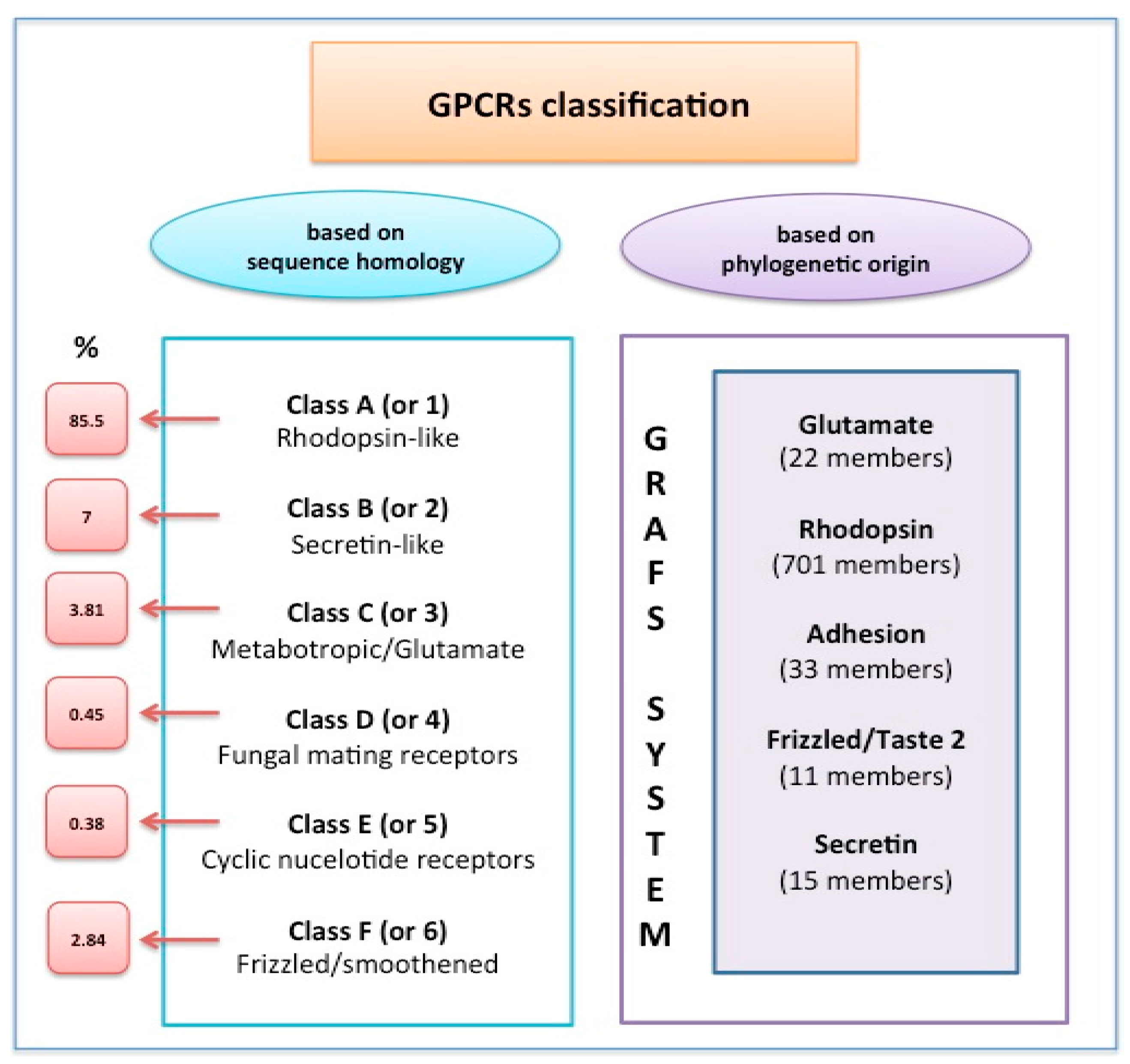
:1. Introduction
2. G Protein-Coupled Receptors (GPCRs) Involved in Early Vascular System Development and Maintenance: The Road to Cell Fate Commitment through Lineage Restriction
2.1. Lysophospholipid Receptors: Angiogenic Actions Mediated by SPHINGOSINE 1P Receptors
2.2. Thrombin Receptors
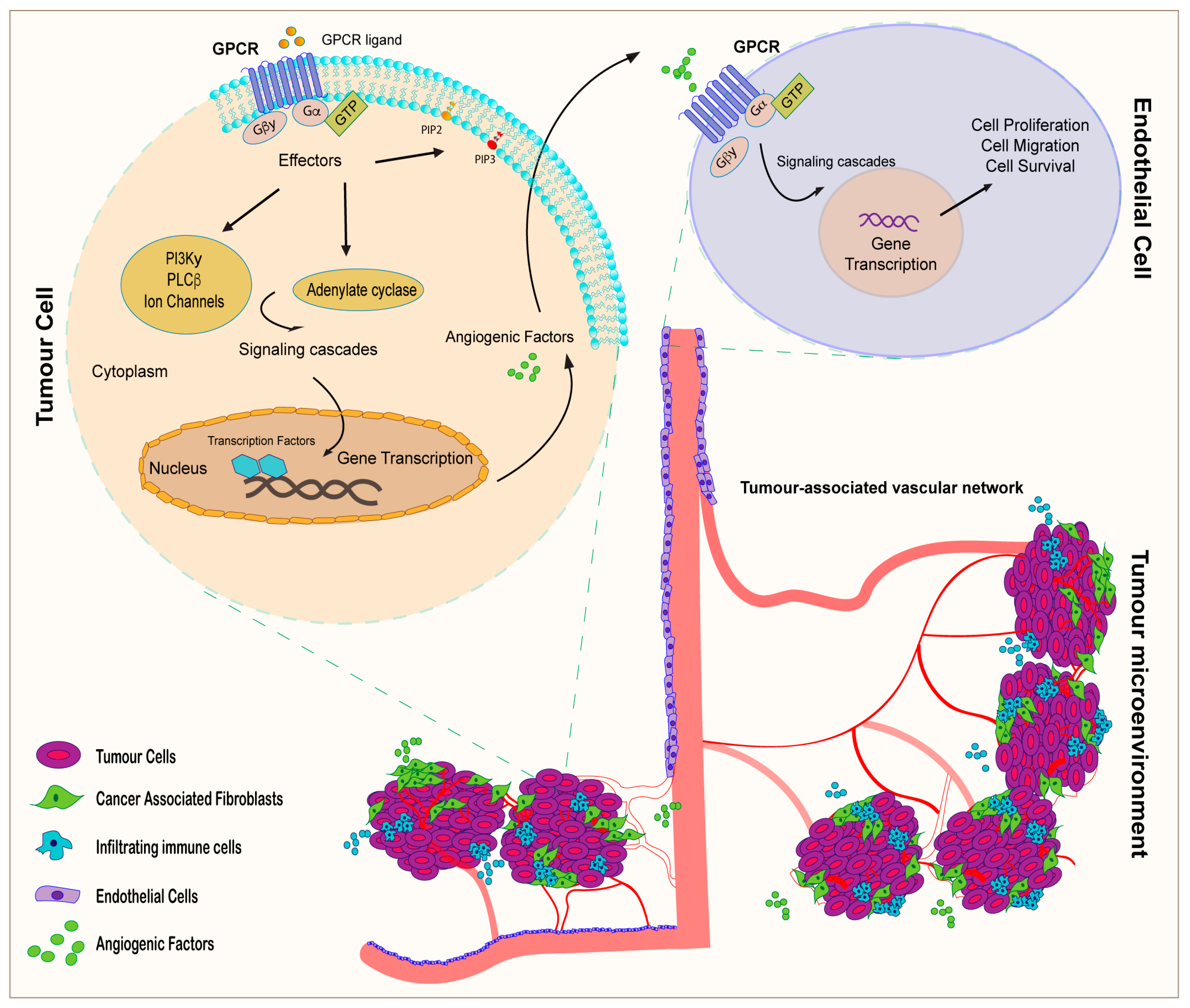
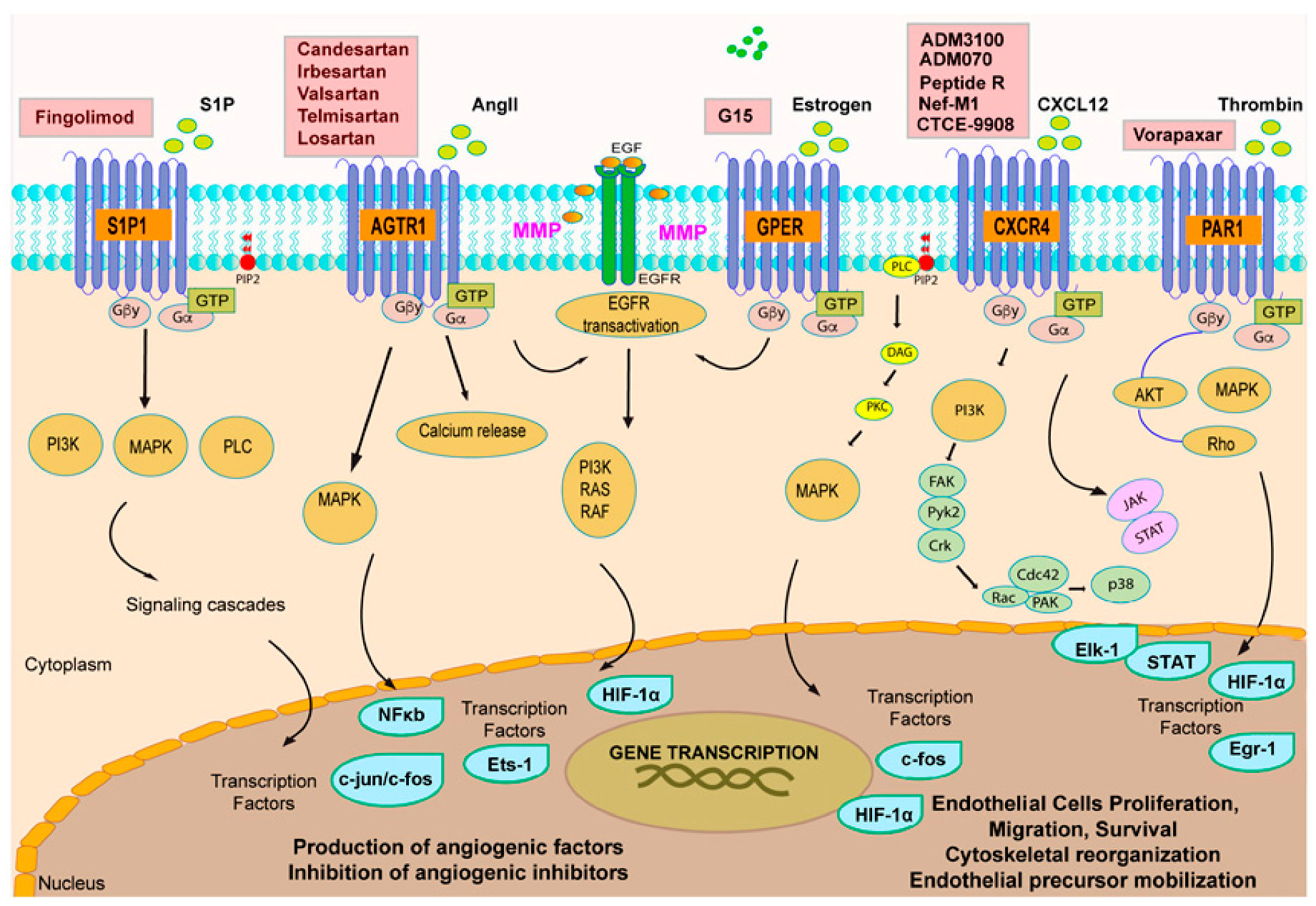
3. GPCR Signaling and Tumor Angiogenesis: An Interactive Loop Promoting Disease Progression
3.1. Chemokine Receptors CXCR4 and CXCR5: A Signaling Hub in Tumor Angiogenesis
3.2. Orphan GPCRs and Tumor Angiogenesis: The Case of GPER
4. Concluding Remarks and Challenges Ahead
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GPCRs | G protein-Coupled Receptors |
| 7TMRs | 7 Transmembrane Receptors |
| GTP | Guanosine 5′-Triphosphate |
| VEGF | Vascular Endothelial Growth Factor |
| HIF-1 | Hypoxia Inducible Factor 1 |
| SDF-1 | Stromal Derived Factor-1 |
| S1P | Sphingosine 1-Phosphate |
| ELDT1 | Epidermal Growth Factor, Latrophilin and Seven Transmembrane Domain-Containing Protein-1 |
| GPER | G Protein Estrogen Receptor |
| PARs | Protease-Activated Receptors |
| LPARs | Lysophosphatidic Acid Receptors |
| AGTR | Angiotensin Receptor |
| VEGFR2 | Vascular Endothelial Growth Factor Receptor 2 |
| ROS | Reactive Oxygen Species |
| ECs | Endothelial Cells |
| EPCs | Endothelial Progenitor Cells |
| VSMCs | Vascular Smooth Muscle Cells |
| CAFs | Cancer Associated Fibroblasts |
| TAMs | Tumor Associated Macrophages |
| BBB | Blood–Brain Barrier |
References
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, signaling, and physiological functions of G-proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Strange, P.G. Signaling mechanisms of GPCR ligands. Curr. Opin. Drug Discov. Dev. 2008, 11, 196–202. [Google Scholar]
- Ho, M.K.; Su, Y.; Yeung, W.W.; Wong, Y.H. Regulation of transcription factors by heterotrimeric G proteins. Curr. Mol. Pharmacol. 2009, 2, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Jockers, R. Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep. 2006, 7, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, M.; Siara, M.; Sajewicz, W. G protein-coupled receptors: Abnormalities in signal transmission, disease states and pharmacotherapy. Acta Pol. Pharm. 2014, 71, 229–243. [Google Scholar] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. GPCRs and cancer. Acta Pharmacol. Sin. 2012, 33, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ye, R.D. Role of G protein-coupled receptors in inflammation. Acta Pharmacol. Sin. 2012, 33, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Burnham, W.M.; Cole, D.E. The G protein-coupled receptors: Pharmacogenetics and disease. Crit. Rev. Clin. Lab. Sci. 2005, 42, 311–392. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, C. G protein-coupled receptors and their signaling pathways: Classical therapeutical targets susceptible to novel therapeutic concepts. Curr. Pharm. Des. 2004, 10, 1937–1958. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.X.; Li, W.; Xu, T.R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Moccia, F.; Li, J.H.; Micera, A.; Kyriakides, T.R. Angiogenesis and vasculogenesis in health and disease. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS 2005, 94, 209–231. [Google Scholar]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Vouret-Craviari, V.; Pouysségur, J. Angiogenesis and G-protein-coupled receptors: Signals that bridge the gap. Oncogene 2001, 20, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Rigiracciolo, D.; De Marco, P.; Avino, S.; Cappello, A.R.; Rosano, C.; Maggiolini, M.; De Francesco, E.M. Recent Advances on the Role of G Protein-Coupled Receptors in Hypoxia-Mediated Signaling. AAPS J. 2016, 18, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Demory, A.; Peyre, F.; Kroll, J.; Augustin, H.G.; Helfrich, W.; Kzhyshkowska, J.; Schledzewski, K.; Arnold, B.; Goerdt, S. Wnt2 acts as a cell type-specific, autocrine growth factor in rat hepatic sinusoidal endothelial cells cross-stimulating the VEGF pathway. Hepatology 2008, 47, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Barcelos, L.S.; Duplaa, C.; Kränkel, N.; Graiani, G.; Invernici, G.; Katare, R.; Siragusa, M.; Meloni, M.; Campesi, I.; Monica, M.; et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ. Res. 2009, 104, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Bao, X.; Al-Ahmad, A.; Liu, J.; Wu, Y.; Dong, W.; Dunn, K.K.; Shusta, E.V.; Palecek, S.P. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of Wnt signaling. Stem Cell Rep. 2014, 3, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; An, S.; Sassmann, A.; Liu, M.; Mastej, V.; Mittal, M.; Zhang, W.; Hong, Z.; Offermanns, S.; Rehman, J.; et al. PAR1 Scaffolds TGFβRII to Downregulate TGF-β Signaling and Activate ESC Differentiation to Endothelial Cells. Stem Cell Rep. 2016, 7, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Alberelli, M.A.; De Candia, E. Functional role of protease activated receptors in vascular biology. Vasc. Pharmacol. 2014, 62, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.J.; Ishihara, H.; Kahn, M.L.; Farese, R.V., Jr.; Coughlin, S.R. Role of the thrombin receptor in development and evidence for a second receptor. Nature 1996, 381, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Miao, X.; Luan, Y.; Zhu, L.; Kong, F.; Lu, Q.; Pernow, J.; Nilsson, G.; Li, N. PAR1-stimulated platelet releasate promotes angiogenic activities of endothelial progenitor cells more potently than PAR4-stimulated platelet releasate. J. Thromb. Haemost. 2015, 13, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Haralabopoulos, G.C.; Grant, D.S.; Kleinman, H.K.; Maragoudakis, M.E. Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am. J. Physiol. 1997, 273, C239–C245. [Google Scholar] [PubMed]
- Tsopanoglou, N.E.; Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by regulation of its receptors. J. Biol. Chem. 1999, 274, 23969–23976. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Yamashita, T.; Proia, R.L. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 2003, 102, 3665–3667. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wang, Y.; Huang, H.; Yu, W.; Bai, M.; Zhang, L.; Bryan, B.A.; Wang, Y.; Luo, J.; Li, D.; et al. GPR126 protein regulates developmental and pathological angiogenesis through modulation of VEGFR2 receptor signaling. J. Biol. Chem. 2014, 289, 34871–34885. [Google Scholar] [CrossRef] [PubMed]
- Tamama, K.; Kon, J.; Sato, K.; Tomura, H.; Kuwabara, A.; Kimura, T.; Kanda, T.; Ohta, H.; Ui, M.; Kobayashi, I. Extracellular mechanism through the Edg family of receptors might be responsible for sphingosine-1-phosphate-induced regulation of DNA synthesis and migration of rat aortic smooth-muscle cells. Biochem. J. 2001, 353, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Zhang, L.L.; Zhang, L.Z.; Li, W.; Li, M.; Wen, K. Sphingosine 1-Phosphate Postconditioning Protects against Myocardial Ischemia/reperfusion Injury in Rats via Mitochondrial Signaling and Akt-Gsk3β Phosphorylation. Arch. Med. Res. 2017, 48, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Sakamoto, T.; Robador, P.A.; Tomita, K.; Levi, R. S1P receptor 1-Mediated Anti-Renin-Angiotensin System Cardioprotection: Pivotal Role of Mast Cell Aldehyde Dehydrogenase Type 2. J. Pharmacol. Exp. Ther. 2017, 362, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Skoura, A.; Sanchez, T.; Claffey, K.; Mandala, S.M.; Proia, R.L.; Hla, T. Essential role of sphingosine 1-phosphate receptor 2 in pathological angiogenesis of the mouse retina. J. Clin. Investig. 2007, 117, 2506–2516. [Google Scholar] [CrossRef] [PubMed]
- Olszewska-Pazdrak, B.; Carney, D.H. Systemic administration of thrombin peptide TP508 enhances VEGF-stimulated angiogenesis and attenuates effects of chronic hypoxia. J. Vasc. Res. 2013, 50, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Mitsos, S.; Koletsis, E.N.; Katsanos, K.; Bravou, V.; Kolonitsiou, F.; Marinos, E.; Flordellis, C.S.; Dougenis, D. Intramyocardial thrombin promotes angiogenesis and improves cardiac function in an experimental rabbit model of acute myocardial infarction. J. Thorac. Cardiovasc. Surg. 2014, 147, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Alam, K.; Nawaz, M.I.; Mohammad, G.; Van den Eynde, K.; Siddiquei, M.M.; Mousa, A.; De Hertogh, G.; Opdenakker, G. Upregulation of Thrombin/Matrix Metalloproteinase-1/Protease-Activated Receptor-1 Chain in Proliferative Diabetic Retinopathy. Curr. Eye Res. 2016, 41, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Prossnitz, E.R.; Barton, M. GPER/GPR30 and Regulation of Vascular Tone and Blood Pressure. Immunol. Endocr. Metab. Agents Med. Chem. 2011, 11, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Tropea, T.; De Francesco, E.M.; Rigiracciolo, D.; Maggiolini, M.; Wareing, M.; Osol, G.; Mandalà, M. Pregnancy Augments G Protein Estrogen Receptor (GPER) Induced Vasodilation in Rat Uterine Arteries via the Nitric Oxide-cGMP Signaling Pathway. PLoS ONE 2015, 10, e0141997. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Angelone, T.; Pasqua, T.; Pupo, M.; Cerra, M.C.; Maggiolini, M. GPER mediates cardiotropic effects in spontaneously hypertensive rat hearts. PLoS ONE 2013, 8, e69322. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, J.; Oyesanya, R.A.; Lee, Z.; Mukherjee, A.; Fang, X. Sp-1 and c-Myc mediate lysophosphatidic acid-induced expression of vascular endothelial growth factor in ovarian cancer cells via a hypoxia-inducible factor-1-independent mechanism. Clin. Cancer Res. 2009, 15, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Kim, J.Y.; Cho, Y.; An, H.; Lee, N.; Jo, H.; Ban, C.; Seo, J.H. Overexpression of angiotensin II type 1 receptor in breast cancer cells induces epithelial-mesenchymal transition and promotes tumor growth and angiogenesis. Biochim. Biophys. Acta 2016, 1863, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Egami, K.; Murohara, T.; Shimada, T.; Sasaki, K.; Shintani, S.; Sugaya, T.; Ishii, M.; Akagi, T.; Ikeda, H.; Matsuishi, T.; et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J. Clin. Investig. 2003, 112, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Sodhi, A.; Pece, S.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001, 61, 2641–2648. [Google Scholar] [PubMed]
- Sodhi, A.; Montaner, S.; Patel, V.; Zohar, M.; Bais, C.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 2000, 60, 4873–4880. [Google Scholar] [PubMed]
- Xue, L.J.; Mao, X.B.; Ren, L.L.; Chum, X.Y. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. 2017, 6, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhong, T. The association of CXCR4 expression with clinicopathological significance and potential drug target in prostate cancer: A meta-analysis and literature review. Drug Des. Dev. Ther. 2015, 9, 5115–5122. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Li, H.Y.; Su, X.L.; Wang, X.Y.; Tian, T.; Li, F.; Ren, G.S. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2010, 29, 31. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lange, A.; Mottram, P.; Alvarez, X.; Cheng, P.; Hogan, M.; Moons, L.; Wei, S.; Zou, L.; Machelon, V.; et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005, 65, 465–472. [Google Scholar] [PubMed]
- Rupertus, K.; Sinistra, J.; Scheuer, C.; Nickels, R.M.; Schilling, M.K.; Menger, M.D.; Kollmar, O. Interaction of the chemokines I-TAC (CXCL11) and SDF-1 (CXCL12) in the regulation of tumor angiogenesis of colorectal cancer. Clin. Exp. Metastasis 2014, 31, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.C.; Chen, J.H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.T.; Huang, Y.L.; Tzeng, H.E.; Tsai, C.H.; Wang, S.W.; Tang, C.H. CCL5 promotes vascular endothelial growth factor expression and induces angiogenesis by down-regulating miR-199a in human chondrosarcoma cells. Cancer Lett. 2015, 357, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Margariti, A.; Kelaini, S.; Cochrane, A.; Guha, S.T.; Hu, Y.; Stitt, A.W.; Zhang, L.; Xu, Q. MicroRNA-199b modulates vascular cell fate during iPS cell differentiation by targeting the Notch ligand Jagged1 and enhancing VEGF signalling. Stem Cells 2015, 33, 1405–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sax, M.J.; Gasch, C.; Athota, V.R.; Freeman, R.; Rasighaemi, P.; Westcott, D.E.; Day, C.J.; Nikolic, I.; Elsworth, B.; Wei, M.; et al. Cancer cell CCL5 mediates bone marrow independent angiogenesis in breast cancer. Oncotarget 2016, 7, 85437–85449. [Google Scholar] [PubMed]
- Wang, Y.; Cho, S.G.; Wu, X.; Siwko, S.; Liu, M. G-protein coupled receptor 124 (GPR124) in endothelial cells regulates vascular endothelial growth factor (VEGF)-induced tumor angiogenesis. Curr. Mol. Med. 2014, 14, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Masiero, M.; Simões, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Favara, D.M.; Banham, A.H.; Harris, A.L. A review of ELTD1, a pro-angiogenic adhesion GPCR. Biochem. Soc. Trans. 2014, 42, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Serban, F.; Daianu, O.; Tataranu, L.G.; Artene, S.A.; Emami, G.; Georgescu, A.M.; Alexandru, O.; Purcaru, S.O.; Tache, D.E.; Danciulescu, M.M.; et al. Silencing of epidermal growth factor, latrophilin and seven transmembrane domain-containing protein 1 (ELTD1) via siRNA-induced cell death in glioblastoma. J. Immunoass. Immunochem. 2017, 38, 21–33. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [PubMed]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.R.; Weinstein, B.M. Arterial-venous specification during development. Circ. Res. 2009, 104, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Goldie, L.C.; Nix, M.K.; Hirschi, K.K. Embryonic vasculogenesis and hematopoietic specification. Organogenesis 2008, 4, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Iruela-Arispe, M.L.; Davis, G.E. Cellular and molecular mechanisms of vascular lumen formation. Dev. Cell 2009, 16, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Eichmann, A. Axon guidance molecules in vascular patterning. Cold Spring Harb. Perspect. Biol. 2010, 2, a001875. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Dolatshad, N.F.; Hellen, N.; Jabbour, R.J.; Harding, S.E.; Földes, G. G-protein Coupled Receptor Signaling in Pluripotent Stem Cell-derived Cardiovascular Cells: Implications for Disease Modeling. Front. Cell Dev. Biol. 2015, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Stenman, J.M.; Rajagopal, J.; Carroll, T.J.; Ishibashi, M.; McMahon, J.; McMahon, A.P. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 2008, 322, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Paes, K.T.; Wang, E.; Henze, K.; Vogel, P.; Read, R.; Suwanichkul, A.; Kirkpatrick, L.L.; Potter, D.; Newhouse, M.M.; Rice, D.S. Frizzled 4 is required for retinal angiogenesis and maintenance of the blood-retina barrier. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6452–6461. [Google Scholar] [CrossRef] [PubMed]
- Peghaire, C.; Bats, M.L.; Sewduth, R.; Jeanningros, S.; Jaspard, B.; Couffinhal, T.; Duplàa, C.; Dufourcq, P. Fzd7 (Frizzled-7) Expressed by Endothelial Cells Controls Blood Vessel Formation Through Wnt/β-Catenin Canonical Signaling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.C.; Kimura, K.; Nagano, M.; Yamashita, T.; Ohneda, K.; Sugimori, H.; Sato, F.; Sakakibara, Y.; Hamada, H.; Yoshikawa, H.; et al. Identification of human placenta-derived mesenchymal stem cells involved in re-endothelialization. J. Cell. Physiol. 2011, 226, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Gering, M.; Patient, R. Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev. Cell 2005, 8, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Byrd, N.; Grabel, L. Hedgehog signaling in murine vasculogenesis and angiogenesis. Trends Cardiovasc. Med. 2004, 14, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Renault, M.A.; Robbesyn, F.; Chapouly, C.; Yao, Q.; Vandierdonck, S.; Reynaud, A.; Belloc, I.; Traiffort, E.; Ruat, M.; Desgranges, C.; et al. Hedgehog-dependent regulation of angiogenesis and myogenesis is impaired in aged mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.N.; Koudijs, M.J.; Patient, R.K.; Ingham, P.W.; Schulte-Merker, S.; van Eeden, F.J. Hedgehog signaling via a calcitonin receptor-like receptor can induce arterial differentiation independently of VEGF signaling in zebrafish. Blood 2012, 120, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.G.; Albacker, C.E.; Lu, Y.F.; Jang, I.H.; Lim, Y.; Heffner, G.C.; Arora, N.; Bowman, T.V.; Lin, M.I.; Lensch, M.W. Signaling axis involving Hedgehog, Notch, and Scl promotes the embryonic endothelial-to-hematopoietic transition. Proc. Natl. Acad. Sci. USA 2013, 110, E141–E150. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Barnett, A.; Zhang, Y.; Yu, X.; Luo, Y. Poststroke Sonic Hedgehog Agonist Treatment Improves Functional Recovery by Enhancing Neurogenesis and Angiogenesis. Stroke 2017, 48, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Huang, M.; He, Q.W.; Zhang, Y.; Opoku, E.N.; Yang, H.; Jin, H.J.; Xia, Y.P.; Hu, B. Administration of sonic hedgehog protein induces angiogenesis and has therapeutic effects after stroke in rats. Neuroscience 2017, 352, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Seitz, G.; Boehmler, A.M.; Kanz, L.; Möhle, R. The role of sphingosine 1-phosphate receptors in the trafficking of hematopoietic progenitor cells. Ann. N. Y. Acad. Sci. 2005, 1044, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Xiu, X.; Zhao, Y.; Gui, M. Improved Proliferation and Differentiation of Bone Marrow Mesenchymal Stem Cells Into Vascular Endothelial Cells With Sphingosine 1-Phosphate. Transplant. Proc. 2015, 47, 2035–2040. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T.; et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2006, 439, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Li, N.; Evans, S.M.; Diaz, M.F.; Wenzel, P.L. Biomechanical force in blood development: Extrinsic physical cues drive pro-hematopoietic signaling. Differentiation 2013, 86, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Carvalho, J.; Looso, M.; Singh, P.; Chennupati, R.; Preussner, J.; Günther, S.; Albarrán-Juárez, J.; Tischner, D.; Classen, S.; et al. Single-cell profiling reveals heterogeneity and functional patterning of GPCR expression in the vascular system. Nat. Commun. 2017, 8, 15700. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Bell, B.M. Functions of sphingolipids and sphingolipid breakdown products in cellular regulation. Science 1989, 243, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Merrill, A.H. Spingolipid metabolism and cell growth regulation. FASEB J. 1996, 10, 1388–1397. [Google Scholar] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 114, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as a second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Van Brocklyn, J.R.; Thangada, S.; Liu, C.H.; Hand, A.R.; Menzeleev, R.; Spiegel, S.; Hla, T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 1998, 279, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, Y.; Takuwa, N.; Sugimoto, N. The Edg family G protein-coupled receptors for lysophospholipids: Their signaling properties and biological activities. J. Biochem. 2002, 131, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Van Brocklyn, J.R.; Hobson, J.P.; Movafagh, S.; Zukowska-Grojec, Z.; Milstien, S.; Spiegel, S. Sphingosine 1-phosphate stimulates cell migration through a Gi-coupled cell surface receptor. Potential involvement in angiogenesis. J. Biol. Chem. 1999, 274, 35343–35350. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Watanabe, T.; Sato, K.; Kon, J.; Tomura, H.; Tamama, K.; Kuwabara, A.; Kanda, T.; Kobayashi, I.; Ohta, H.; et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem. J. 2000, 348, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Sharma, S. Mechanism of sphingosine-1-phosphate induced cardioprotection against I/R injury in diabetic rat heart: Possible involvement of glycogen synthase kinase 3β and mitochondrial permeability transition pore. Clin. Exp. Pharmacol. Physiol. 2016, 43, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Oyama, O.; Sugimoto, N.; Qi, X.; Takuwa, N.; Mizugishi, K.; Koizumi, J.; Takuwa, Y. The lysophospholipid mediator sphingosine-1-phosphate promotes angiogenesis in vivo in ischaemic hindlimbs of mice. Cardiovasc. Res. 2008, 78, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Okamoto, Y.; Murakawa, T.; Wang, F.; Oyama, O.; Ohkawa, R.; Yoshioka, K.; Du, W.; Sugimoto, N.; Yatomi, Y.; et al. Sustained delivery of sphingosine-1-phosphate using poly(lactic-co-glycolic acid)-based microparticles stimulates Akt/ERK-eNOS mediated angiogenesis and vascular maturation restoring blood flow in ischemic limbs of mice. Eur. J. Pharmacol. 2010, 634, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Dobierzewska, A.; Palominos, M.; Sanchez, M.; Dyhr, M.; Helgert, K.; Venegas-Araneda, P.; Tong, S.; Illanes, S.E. Impairment of Angiogenic Sphingosine Kinase-1/Sphingosine-1-Phosphate Receptors Pathway in Preeclampsia. PLoS ONE 2016, 11, e0157221. [Google Scholar] [CrossRef] [PubMed]
- Brait, V.H.; Tarrasón, G.; Gavaldà, A.; Godessart, N.; Planas, A.M. Selective Sphingosine 1-Phosphate Receptor 1 Agonist Is Protective Against Ischemia/Reperfusion in Mice. Stroke 2016, 47, 3053–3056. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhang, D.; Dai, D.; Zhang, W.; Zhang, L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. 2016, 65, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.G.; Choi, J.W. Identification of sphingosine 1-phosphate receptor subtype 1 (S1P1) as a pathogenic factor in transient focal cerebral ischemia. Mol. Neurobiol. 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guria, K.; Guria, G.T. Spatial aspects of blood coagulation: Two decades of research on the self-sustained traveling wave of thrombin. Thromb. Res. 2015, 135, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Posma, J.J.; Posthuma, J.J.; Spronk, H.M. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. 2016, 14, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Moers, A.; Nieswandt, B.; Massberg, S.; Wettschureck, N.; Grüner, S.; Konrad, I.; Schulte, V.; Aktas, B.; Gratacap, M.P.; Simon, M.I.; et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 2003, 9, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Van den Eshof, B.L.; Hoogendijk, A.J.; Simpson, P.J.; van Alphen, F.P.J.; Zanivan, S.; Mertens, K.; Meijer, A.B.; van den Biggelaar, M. Paradigm of biased PAR1 (Protease-Activated Receptor-1) activation and inhibition in endothelial cells dissected by phosphoproteomics. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, E.; Habib, A.; Lebret, M.; Yang, R.; Levy-Toledano, S.; Tobelem, G. Thrombin induces angiogenesis and vascular endothelial growth factor expression in human endothelial cells: Possible relevance to HIF-1α. Thromb. Haemost. 2003, 1, 1096–1102. [Google Scholar] [CrossRef]
- Caunt, M.; Huang, Y.Q.; Brooks, P.C.; Karpatkin, S. Thrombin induces neoangiogenesis in the chick chorioallantoic membrane. J. Thromb. Haemost. 2003, 1, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, P.; Salo, T.; Kalluri, R. Tumor microenvironment and angiogenesis. Front. Biosci. 2008, 13, 6537–6553. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 5, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Tan, H.S.; Datta, A.; Lai, R.C.; Zhang, H.; Meng, W.; Lim, S.K.; Sze, S.K. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol. Cell. Proteom. 2010, 9, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, R.M.; D’Amore, P.A. Transcriptional regulation of vascular endothelial growth factor in cancer. Cytokine Growth Factor Rev. 2005, 16, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Shimoda, L.A.; Prabhakar, N.R. Regulation of gene expression by HIF-1. Novartis Found. Symp. 2006, 272, 2–8. [Google Scholar] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Schito, L.; Wouters, B.G.; Eliasof, S.; Kerbel, R.S. Targeting Hypoxia-Inducible Factors for Antiangiogenic Cancer Therapy. Trends Cancer 2017, 3, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. Pharmacotherapeutic Targeting of G Protein-Coupled Receptors in Oncology: Examples of Approved Therapies and Emerging Concepts. Drugs 2017, 77, 951–965. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Meshram, R.J. Targeting tumor micro-environment for design and development of novel anti-angiogenic agents arresting tumor growth. Prog. Biophys. Mol. Biol. 2013, 113, 333–354. [Google Scholar] [CrossRef] [PubMed]
- Kanehira, M.; Fujiwara, T.; Nakajima, S.; Okitsu, Y.; Onishi, Y.; Fukuhara, N.; Ichinohasama, R.; Okada, Y.; Harigae, H. An lysophosphatidic acid receptors 1 and 3 axis governs cellular senescence of mesenchymal stromal cells and promotes growth and vascularization of multiple myeloma. Stem Cells 2017, 35, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Arriazu, R.; Durán, E.; Pozuelo, J.M.; Santamaria, L. Expression of lysophosphatidic acid receptor 1 and relation with cell proliferation, apoptosis, and angiogenesis on preneoplastic changes induced by cadmium chloride in the rat ventral prostate. PLoS ONE 2013, 8, e57742. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Prestwich, G.D. Inhibition of tumor growth and angiogenesis by a lysophosphatidic acid antagonist in an engineered three-dimensional lung cancer xenograft model. Cancer 2010, 116, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Ateeq, B.; Cao, Q.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Kalyana-Sundaram, S.; Lonigro, R.J.; Helgeson, B.E.; Bhojani, M.S.; et al. AGTR1over-expressiondefinesa subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10284–10289. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.A.; Choi, C.H.; Do, I.G.; Song, S.Y.; Lee, J.K.; Cho, Y.J.; Choi, J.J.; Jeon, H.K.; Ryu, J.Y.; Lee, Y.Y.; et al. Dual targeting of angiotensin receptors (AGTR1 and AGTR2) in epithelial ovarian carcinoma. Gynecol. Oncol. 2014, 135, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.S.; Paik, J.H.; Furneaux, H.; Hla, T. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J. Clin. Investig. 2004, 114, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Maczis, M.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate and estrogen signaling in breast cancer. Adv. Biol. Regul. 2016, 60, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Kim, R.H.; Allegood, J.C.; Mitra, P.; Ramachandran, S.; Nagahashi, M.; Harikumar, K.B.; Hait, N.C.; Milstien, S.; Spiegel, S. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 2010, 285, 10477–10486. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Maceyka, M.; Hait, N.C.; Paugh, S.W.; Sankala, H.; Milstien, S.; Spiegel, S. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005, 579, 5313–5317. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Horie, S.; Muto, S.; Otsuki, Y.; Matsumoto, K.; Morimoto, J.; Gotoh, R.; Okuyama, A.; Suzuki, S.; Katsuoka, Y.; et al. Selective cancer cell apoptosis induced by FTY720; Evidence for a Bcl-dependent pathway and impairment in ERK activity. Anticancer Res. 2003, 23, 3183–3193. [Google Scholar] [PubMed]
- LaMontagne, K.; Littlewood-Evans, A.; Schnell, C.; O’Reilly, T.; Wyder, L.; Sanchez, T.; Probst, B.; Butler, J.; Wood, A.; Liau, G.; et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006, 66, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Schmid, G.; Guba, M.; Papyan, A.; Ischenko, I.; Bruckel, M.; Bruns, C.J.; Jauch, K.W.; Graeb, C. FTY720 inhibits tumor growth and angiogenesis. Transplant. Proc. 2005, 37, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, Y.; Mollard, S.; Faucher-Durand, K.; Richard, L.; Nizou, A.; Cook-Moreau, J.; Baaj, Y.; Qiu, H.; Plainard, X.; Fourcade, L.; et al. Fingolimod potentiates the effects of sunitinib malate in a rat breast cancer model. Breast Cancer Res. Treat. 2012, 134, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell Res. 2011, 317, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2001, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Smadja, D.M.; Bieche, I.; Uzan, G.; Bompais, H.; Muller, L.; Boisson-Vidal, C.; Vidaud, M.; Aiach, M.; Gaussem, P. PAR-1 activation on human late endothelial progenitor cells enhances angiogenesis in vitro with upregulation of the SDF-1/CXCR4 system Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Shih, YT.; Wang, M.C.; Peng, H.H.; Chen, T.F.; Chen, L.; Chang, J.Y.; Chiu, J.J. Modulation of chemotactic and pro-inflammatory activities of endothelial progenitor cells by hepatocellular carcinoma. Cell. Signal. 2012, 24, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.C.; Kim, W.; Son, K.N.; Han, K.Y.; Lee, K.H.; Kleinman, H.K.; Ko, J.; Na, D.S.; Kwon, B.S.; Gho, Y.S.; et al. Angiogenic activity of human CC chemokine CCL15 in vitro and in vivo. FEBS Lett. 2004, 570, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Strasly, M.; Doronzo, G.; Cappello, P.; Valdembri, D.; Arese, M.; Mitola, S.; Moore, P.; Alessandri, G.; Giovarelli, M.; Bussolino, F. CCL16 activates an angiogenic program in vascular endothelial cells. Blood 2004, 103, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Son, K.N.; Kim, C.W.; Ko, J.; Na, D.S.; Kwon, B.S.; Gho, Y.S.; Kim, J. Human CC chemokine CCL23, a ligand for CCR1, induces endothelial cell migration and promotes angiogenesis. Cytokine 2005, 30, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Ridiandries, A.; Tan, J.T.; Bursill, C.A. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1856. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Wang, C.J.; Lin, T.Y.; Hsiao, C.L.; Luo, C.W. CXCL17, an orphan chemokine, acts as a novel angiogenic and anti-inflammatory factor. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E32–E40. [Google Scholar] [CrossRef] [PubMed]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in tumor-promoting inflammation: Impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia 2008, 10, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.Y.; Matsushima, K.; Baba, T.; Mukaida, N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J. Immunol. 2008, 81, 6384–6393. [Google Scholar] [CrossRef]
- Varney, M.L.; Olsen, K.J.; Mosley, R.L.; Singh, R.K. Paracrine regulation of vascular endothelial growth factor—A expression during macrophage-melanoma cell interaction: Role of monocyte chemotactic protein-1 and macrophage colony-stimulating factor. J. Interferon Cytokine Res. 2005, 25, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.Y.; Tsai, H.C.; Chou, P.Y.; Wang, S.W.; Chen, H.T.; Lin, Y.M.; Chiang, I.P.; Chang, T.M.; Hsu, S.K.; Chou, M.C.; et al. CCL3 promotes angiogenesis by dysregulation of miR-374b/VEGF-A axis in human osteosarcoma cells. Oncotarget 2016, 7, 4310–4325. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.T.; Chen, H.T.; Tsou, H.K.; Tan, T.W.; Fong, Y.C.; Chen, P.C.; Yang, W.H.; Wang, S.W.; Chen, J.C.; Tang, C.H. CCL5 promotes VEGF-dependent angiogenesis by down-regulating miR-200b through PI3K/Akt signaling pathway in human chondrosarcoma cells. Oncotarget 2014, 5, 10718–10731. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Liu, S.C.; Sun, H.L.; Huang, T.Y.; Chan, C.H.; Yang, C.Y.; Yeh, H.I.; Huang, Y.L.; Chou, W.Y.; Lin, Y.M.; et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2015, 36, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Bleul, C.C.; Farzan, M.; Choe, H.; Parolin, C.; Clark-Lewis, I.; Sodroski, J.; Springer, T.A. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996, 382, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, H.; Papanastasiou, A.D.; Sirinian, C.; Scopa, C.; Kalofonos, H.P.; Leotsinidis, M.; Papadaki, H. Expression patterns of SDF1/CXCR4 in human invasive breast carcinoma and adjacent normal stroma: Correlation with tumor clinicopathological parameters and patient survival. Pathol. Res. Pract. 2014, 210, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Dewar, A.L.; Farrugia, A.N.; Horvath, N.; Gronthos, S.; To, L.B.; Zannettino, A.C. Tumor angiogenesis is associated with plasma levels of stromal-derived factor-1α in patients with multiple myeloma. Clin. Cancer Res. 2006, 12, 6973–6977. [Google Scholar] [CrossRef] [PubMed]
- Ponomaryov, T.; Peled, A.; Petit, I.; Taichman, R.S.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Magerus, A.; Caruz, A.; Fujii, N.; et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J. Clin. Investig. 2000, 106, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.; Cohen, K.S.; Klein, R.J.; Scadden, D.T.; Chiocca, E.A. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006, 66, 9054–9064. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.X.; Chen, J.H.; Jiang, X.F.; Wang, Q.L.; Chen, Z.Q.; Xhao, W.; Feng, Y.H.; Xin, R.; Shi, J.Q.; Bian, X.W. Activation of chemokine receptor CXCR4 in malignant glioma cells promotes the production of vascular endothelial growth factor. Biochem. Biophys. Res. Commun. 2005, 335, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Kim, H.Y.; Song, I.C.; Yun, H.J.; Jo, D.Y.; Kim, S.; Lee, H.J. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1α. Oncol. Rep. 2012, 28, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.Y.; Soloviev, I.; Chang, P.; Lee, J.; Huang, X.; Zhong, C.; Ferrara, N.; Polakis, P.; Sakanaka, C. Stromal cell-derived factor-1/CXCL12 contributes to MMTV-Wnt1 tumor growth involving Gr1+CD11b+ cells. PLoS ONE 2010, 5, e8611. [Google Scholar] [CrossRef] [PubMed]
- Schols, D.; Struyf, S.; Van Damme, J.; Este, J.A.; Henson, G.; De Clercq, E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J. Exp. Med. 1997, 186, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Donzella, G.A.; Schols, D.; Lin, S.W.; Esté, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; De Clercq, E.; et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Bai, H.; Shao, Y.; Arzigian, M.; Janzen, V.; Attar, E.; Xie, Y.; Scadden, D.T.; Wang, Z.Z. Stromal cell-derived factor-1/CXCR4 signaling modifies the capillary-like organization of human embryonic stem cell-derived endothelium in vitro. Stem Cells 2007, 25, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.M.; Capoccia, B.J.; Devine, S.M.; Dipersio, J.; Trinkaus, K.M.; Ingram, D.; Link, D.C. Angiogenic cells can be rapidly mobilized and efficiently harvested from the blood following treatment with AMD3100. Blood 2006, 108, 3662–3667. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. 2016, 35, 55. [Google Scholar] [CrossRef] [PubMed]
- Katkoori, V.R.; Basson, M.D.; Bond, V.C.; Manne, U.; Bumpers, H.L. Nef-M1, a peptide antagonist of CXCR4, inhibits tumor angiogenesis and epithelial-to-mesenchymal transition in colon and breast cancers. Oncotarget 2015, 6, 27763–27777. [Google Scholar] [CrossRef] [PubMed]
- Porvasnik, S.; Sakamoto, N.; Kusmartsev, S.; Eruslanov, E.; Kim, W.J.; Cao, W.; Urbanek, C.; Wong, D.; Goodison, S.; Rosser, C.J. Effects of CXCR4 antagonist CTCE-9908 on prostate tumor growth. Prostate 2009, 69, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Civelli, O.; Reinscheid, R.K.; Zhang, Y.; Wang, Z.; Fredriksson, R.; Schiöth, H.B. G protein-coupled receptor deorphanizations. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 127–146. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Wojciech, S.; Jockers, R. Hunting for the function of orphan GPCRs—Beyond the search for the endogenous ligand. Br. J. Pharmacol. 2015, 172, 3212–3228. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2017, 176, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: Its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Irving, A.J.; Harvey, J. Emerging roles for the novel estrogen-sensing receptor GPER1 in the CNS. Neuropharmacology 2017, 113, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mårtensson, U.E.; Salehi, S.A.; Windahl, S.; Gomez, M.F.; Swärd, K.; Daszkiewicz-Nilsson, J.; Wendt, A.; Andersson, N.; Hellstrand, P.; Grände, P.O.; et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 2009, 150, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Mauvais-Jarvis, F.; Prossnitz, E.R. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J. Steroid Biochem. Mol. Biol. 2017, 176, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Brunsing, R.L.; Owens, K.S.; Prossnitz, E.R. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing conditions. J. Immunother. 2013, 36, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, A.M.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kashyap, S.; Murphy, B.; Hutson, D.D.; Budish, R.A.; Trimmer, E.H.; Zimmerman, M.A.; Trask, A.J.; Miller, K.S.; Chappell, M.C.; et al. GPER activation ameliorates aortic remodeling induced by salt-sensitive hypertension. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H953–H961. [Google Scholar] [CrossRef] [PubMed]
- Jessup, J.A.; Lindsey, S.H.; Wang, H.; Chappell, M.C.; Groban, L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS ONE 2010, 5, e15433. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Rocca, C.; Scavello, F.; Amelio, D.; Pasqua, T.; Rigiracciolo, D.C.; Scarpelli, A.; Avino, S.; Cirillo, F.; Amodio, N.; et al. Protective role of GPER agonist G-1 on cardiotoxicity induced by doxorubicin. J. Cell. Physiol. 2017, 232, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Gallelli, L.; Perri, P.; De Francesco, E.M.; Rigiracciolo, D.C.; Mastroroberto, P.; Maggiolini, M.; de Franciscis, S. Estrogen receptors and chronic venous disease. Eur. J. Vasc. Endovasc. Surg. 2016, 52, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.D. Heart disease in women: Unappreciated challenges, GPER as a new target. Int. J. Mol. Sci. 2016, 17, E760. [Google Scholar] [CrossRef] [PubMed]
- Barton, M. Not lost in translation: Emerging clinical importance of the G protein-coupled estrogen receptor GPER. Steroids 2016, 111, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Picard, D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol. 2010, 204, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. GPER is involved in the functional liaison between breast tumor cells and cancer-associated fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 2017, 176, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Andò, S.; Maggiolini, M. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1α (HIF-1α) in breast cancer cells and cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef] [PubMed]
- Bartella, V.; De Francesco, E.M.; Perri, M.G.; Curcio, R.; Dolce, V.; Maggiolini, M.; Vivacqua, A. The G protein estrogen receptor (GPER) is regulated by endothelin-1 mediated signaling in cancer cells. Cell Signal. 2016, 28, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.O.; Stephens, N.D.; Qualls, C.R.; Fligelman, T.; Wang, T.; Lin, C.Y.; Burton, E.; Griffith, J.K.; Pollard, J.W. The clinical significance of inflammatory cytokines in primary cell culture in endometrial carcinoma. Mol. Oncol. 2013, 7, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xiong, W.; Li, N.; Liu, H.; He, H.; Du, Y.; Zhang, Z.; Liu, Y. Estrogen stabilizes hypoxia-inducible factor 1α through G protein-coupled estrogen receptor 1 in eutopic endometrium of endometriosis. Fertil. Steril. 2017, 107, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Ferri, N.; Cignarella, A.; Trevisi, L.; Bolego, C. The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1. J. Pharmacol. Exp. Ther. 2017, 361, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Lucki, N.C.; Sewer, M.B. Genistein stimulates MCF-7 breast cancer cell growth by inducing acid ceramidase (ASAH1) gene expression. J. Biol. Chem. 2011, 286, 19399–19409. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jin, X.; Zhao, N.; Ye, X.; Ying, C. Bisphenol A promotes X-linked inhibitor of apoptosis protein-dependent angiogenesis via G protein-coupled estrogen receptor pathway. J. Appl. Toxicol. 2015, 35, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; Avino, S.; De Marco, P.; Bussolati, B.; Maggiolini, M.; De Francesco, E.M. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 2016, 7, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015, 98, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. G-protein coupled receptors: Advances in simulation and drug discovery. Curr. Opin. Struct. Biol. 2016, 41, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, H.; Tanaka, T.; Ohashi, N.; Masuno, H.; Tamamura, H.; Hiramatsu, K.; Araki, T.; Ueda, S.; Oishi, S.; Fujii, N. Therapeutic potential of the chemokine receptor CXCR4 antagonists as multifunctional agents. Biopolymers 2007, 88, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Chan, K.M.; Chiang, T.; Liu, J.Y.; Chern, G.G.; Hsu, F.F.; Wu, Y.H.; Liu, Y.C.; Chen, Y. Dual-functional nanoparticles targeting CXCR4 and delivering antiangiogenic siRNA ameliorate liver fibrosis. Mol. Pharm. 2016, 13, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N. Compensatory angiogenesis and tumor refractoriness. Oncogenesis 2015, 4, e153. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Chiang, T.; Liu, C.H.; Chern, G.G.; Lin, T.T.; Gao, D.Y.; Chen, Y. Delivery of siRNA using CXCR4-targeted nanoparticles modulates tumor microenvironment and achieves a potent antitumor response in liver cancer. Mol. Ther. 2015, 23, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting hypoxic cancer stem cells (CSCs) with doxycycline: Implications for optimizing anti-angiogenic therapy. Oncotarget 2017, 8, 56126–56142. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Wang, J.Y. G Protein-coupled receptor signaling in stem cells and cancer. Int. J. Mol. Sci. 2016, 17, 707. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reybolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| GPCR | Ligand | Target Cell/Tissue | Function | References |
|---|---|---|---|---|
| Frizzled 4, Frizzled 6, Frizzled 8 | Wnt7a, 7b and Wnt2 | ECs | BBB formation, CNS angiogenesis, hepatic angiogenesis | [24,25,26] |
| Frizzled 4, Frizzled 7 | Wnt3A, Norrin | retinal ECs | retinal angiogenesis, BBB formation and maintenance | [27,28] |
| PAR1 | Thrombin | mouse ESCs, human ECs, Platelets | vasculogenesis, angiogenesis | [29,30,31,32,33,34,35,36] |
| S1P1 | S1P | ECs, VSMCs | vasculogenesis, angiogenesis | [37,38,39] |
| GPR126 | Unknown | ECs | proliferation, migration, endothelial tube formation | [40] |
| GPCR | Ligand | Target Cell/Tissue | Pathological Process | Function | References |
|---|---|---|---|---|---|
| S1P1 | S1P | rat aortic smooth muscle cells, rat heart, renin containing mast cells | myocardial ischemia | protection against ischemic injury | [41,42,43] |
| S1P2 | S1P | mouse retinal ECs | retinopathy | release of inflammatory mediators | [44] |
| PAR1 | Thrombin | mouse aorta, ECs | acute myocardial infarction | angiogenesis | [45,46] |
| PAR1 | Thrombin | human retinal microvascular ECs | proliferative diabetic retinopathy | cell proliferation | [47] |
| GPER | Unknown | rat heart | primary and secondary hypertension, myocardial ischemia | regulation of vascular tone and blood pressure | [48,49,50] |
| GPCR | Ligand | Target Cell/Tissue | Function | References |
|---|---|---|---|---|
| LPAR1–LPAR3 | LPA | ovarian cancer cells | activation of HIF/VEGF pathway | [51] |
| AGTR1 | ANGII | TAMs, breast cancer cells | tumor angiogenesis, EMT | [52,53] |
| S1P1 | S1P | TAMs | lymphangiogenesis | [54] |
| KSHV-GPCR | Orphan | ECs | tumor angiogenesis | [55,56] |
| CXCR4–7 | CXCL12 | cancer cells, CAFs | release of angiogenic factors, EPCs activation, angiogenesis | [57,58,59,60,61,62] |
| CXCR5 | CCL5 | chondrosarcoma cells, breast cancer cells | ECs differentiation, release of angiogenic factor | [63,64,65] |
| GPR124 | Orphan | ECs | tumor angiogenesis | [66] |
| ELDT1 | Orphan | tumor associated ECs | tumor angiogenesis | [67,68,69] |
| GPER | Orphan, 17β-Estradiol | breast cancer cells, CAFs | activation of HIF/VEGF pathway, tumor angiogenesis | [70,71,72] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Francesco, E.M.; Sotgia, F.; Clarke, R.B.; Lisanti, M.P.; Maggiolini, M. G Protein-Coupled Receptors at the Crossroad between Physiologic and Pathologic Angiogenesis: Old Paradigms and Emerging Concepts. Int. J. Mol. Sci. 2017, 18, 2713. https://doi.org/10.3390/ijms18122713
De Francesco EM, Sotgia F, Clarke RB, Lisanti MP, Maggiolini M. G Protein-Coupled Receptors at the Crossroad between Physiologic and Pathologic Angiogenesis: Old Paradigms and Emerging Concepts. International Journal of Molecular Sciences. 2017; 18(12):2713. https://doi.org/10.3390/ijms18122713
Chicago/Turabian StyleDe Francesco, Ernestina M., Federica Sotgia, Robert B. Clarke, Michael P. Lisanti, and Marcello Maggiolini. 2017. "G Protein-Coupled Receptors at the Crossroad between Physiologic and Pathologic Angiogenesis: Old Paradigms and Emerging Concepts" International Journal of Molecular Sciences 18, no. 12: 2713. https://doi.org/10.3390/ijms18122713
APA StyleDe Francesco, E. M., Sotgia, F., Clarke, R. B., Lisanti, M. P., & Maggiolini, M. (2017). G Protein-Coupled Receptors at the Crossroad between Physiologic and Pathologic Angiogenesis: Old Paradigms and Emerging Concepts. International Journal of Molecular Sciences, 18(12), 2713. https://doi.org/10.3390/ijms18122713