
Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells

Abstract
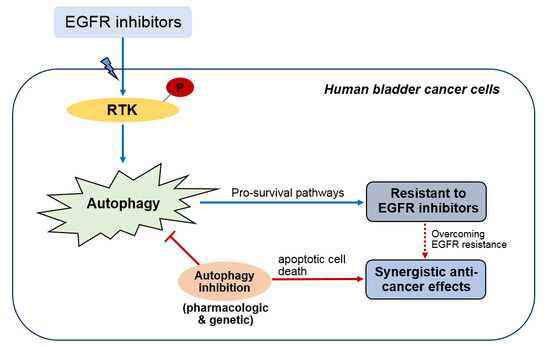
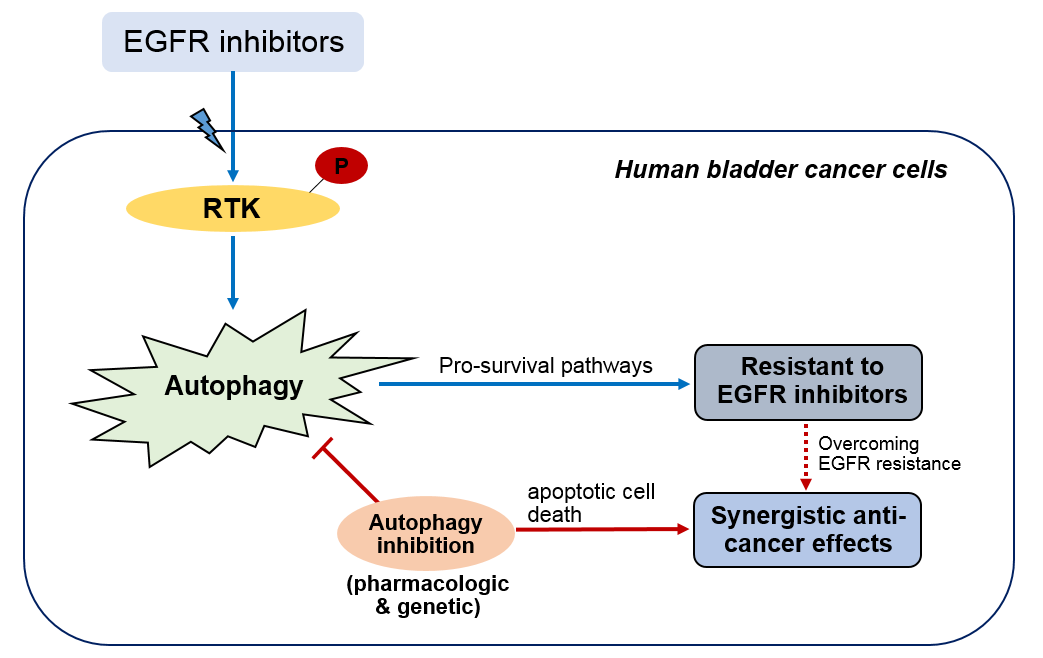
:
1. Introduction
2. Results
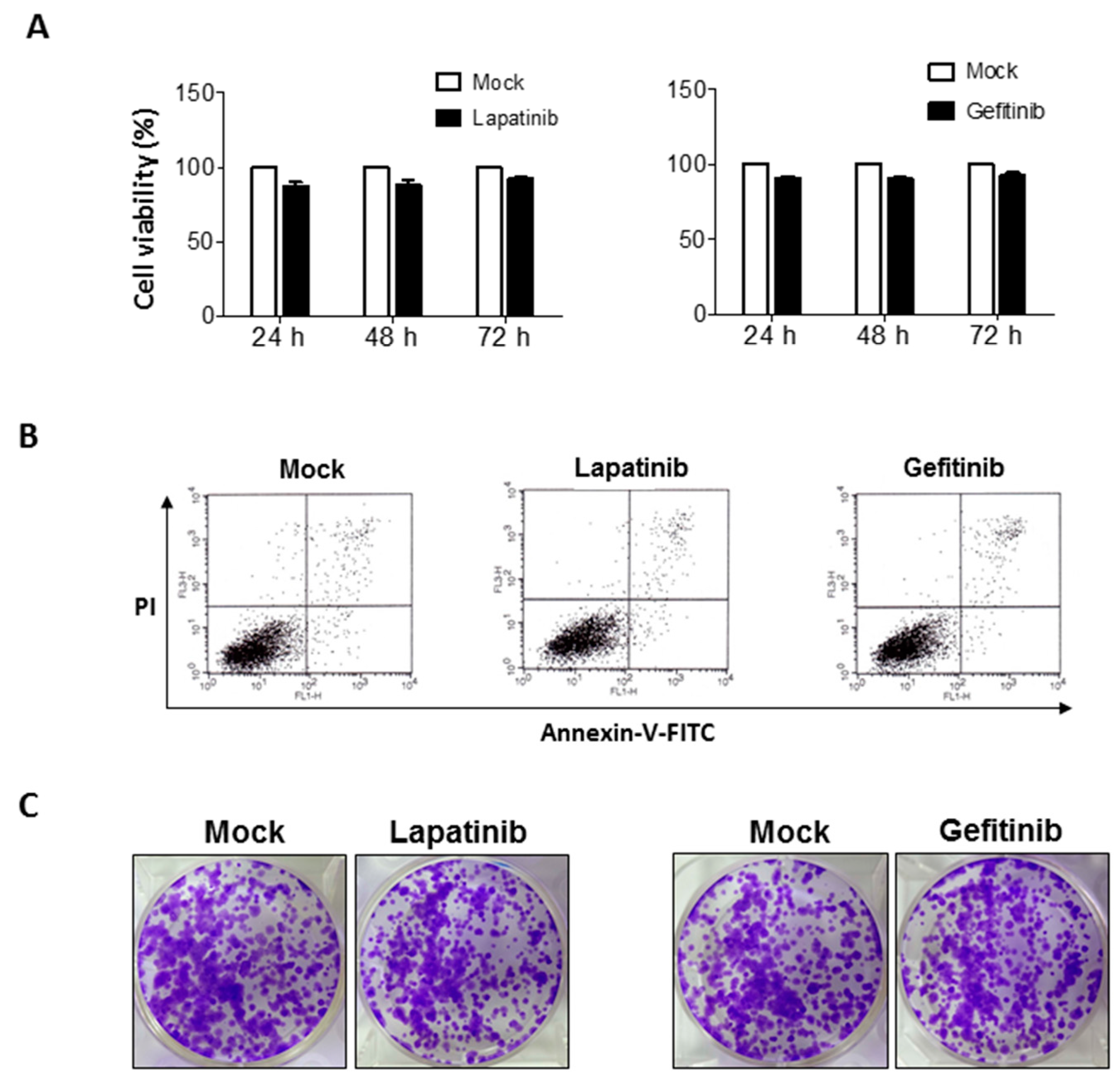
2.1. Non-Significant Anti-Cancer Effects of Epithelial Growth Factor Receptor (EGFR) Inhibitors on Human Bladder Cancer Cells
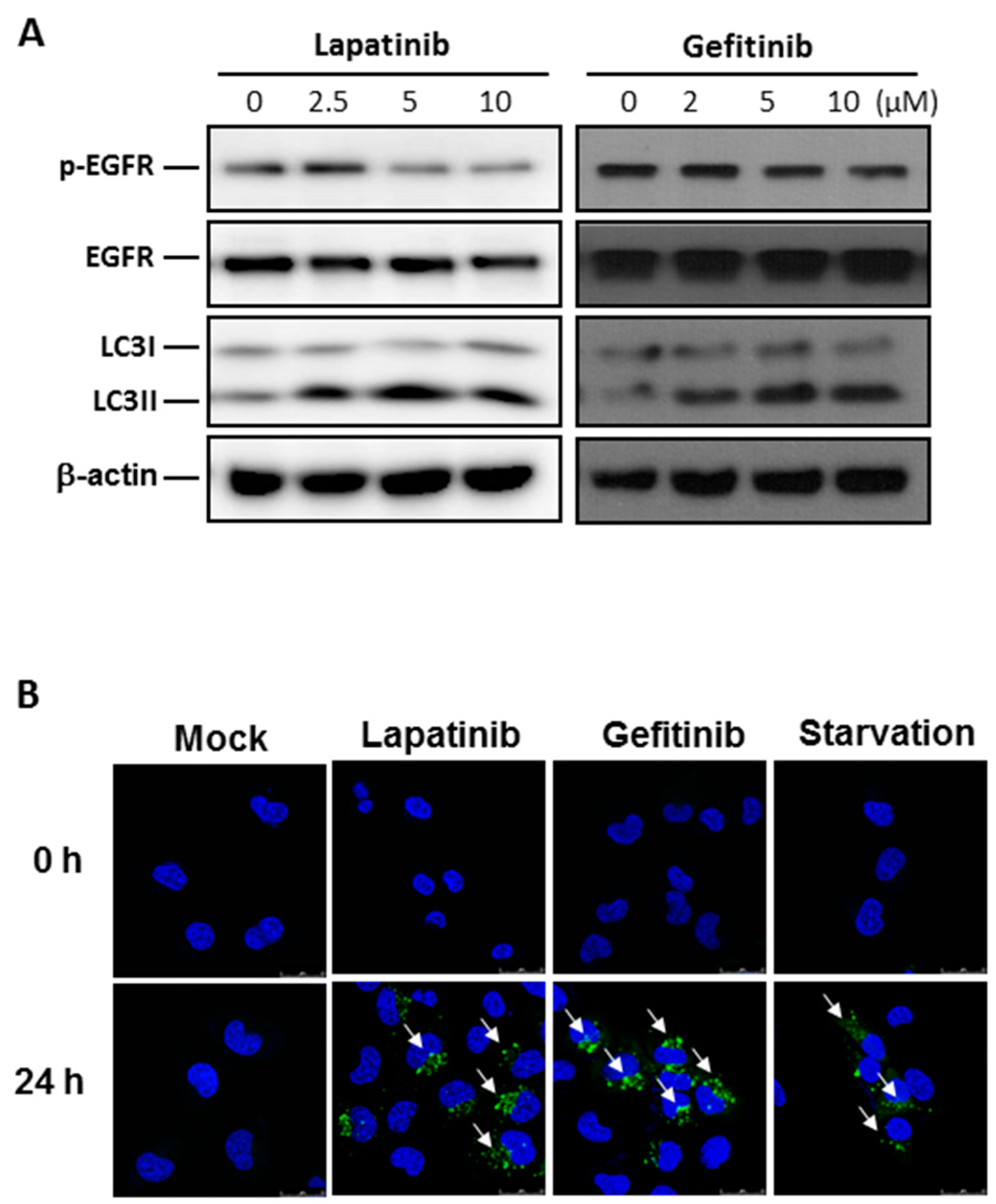
2.2. Autophagy Activation by EGFR Inhibitors in Human Bladder Cancer Cells
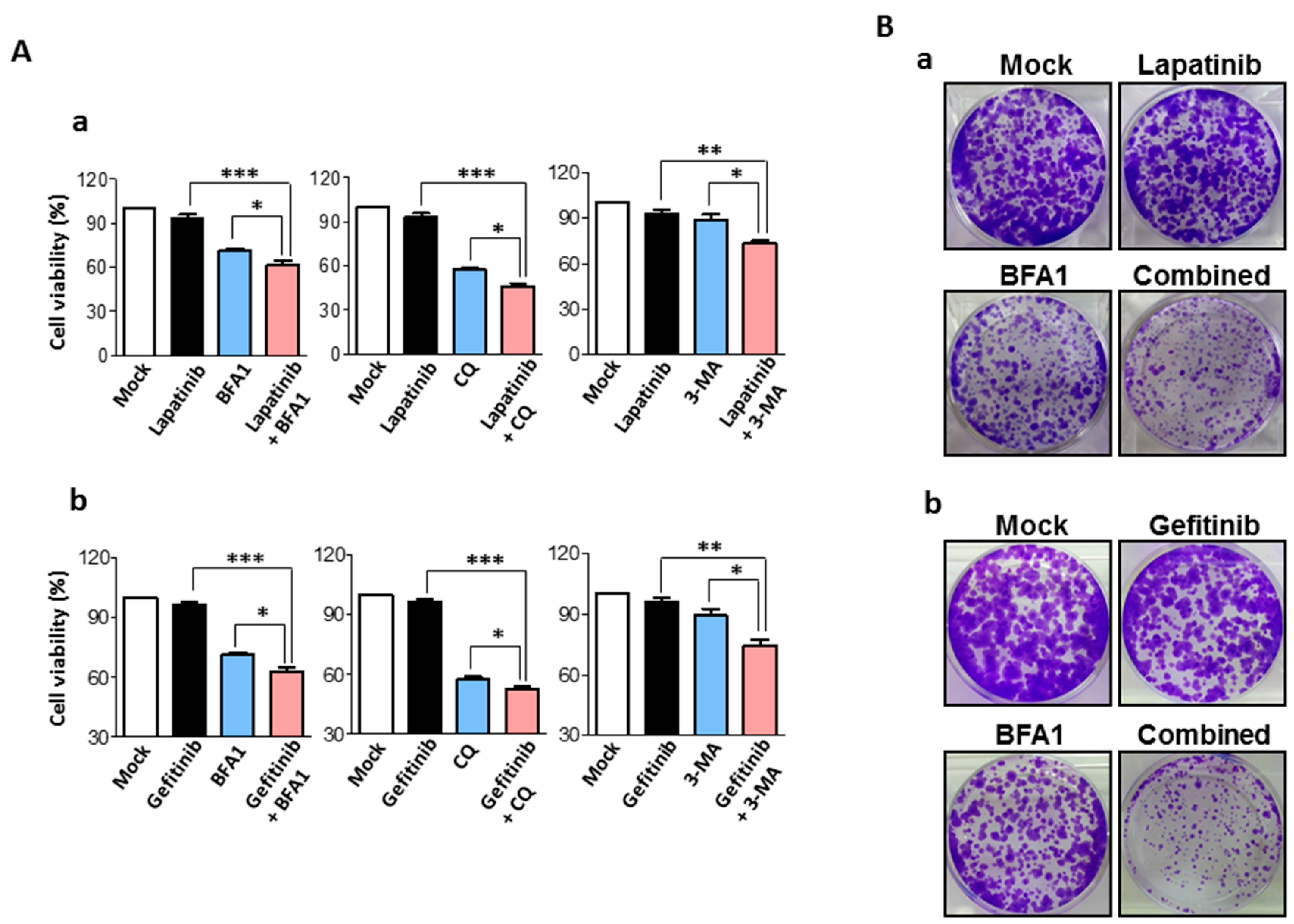
2.3. Synergistic Anti-Cancer Effects by Concurrent Treatment of EGFR Inhibitors and Autophagy Inhibitors in Human Bladder Cancer Cells
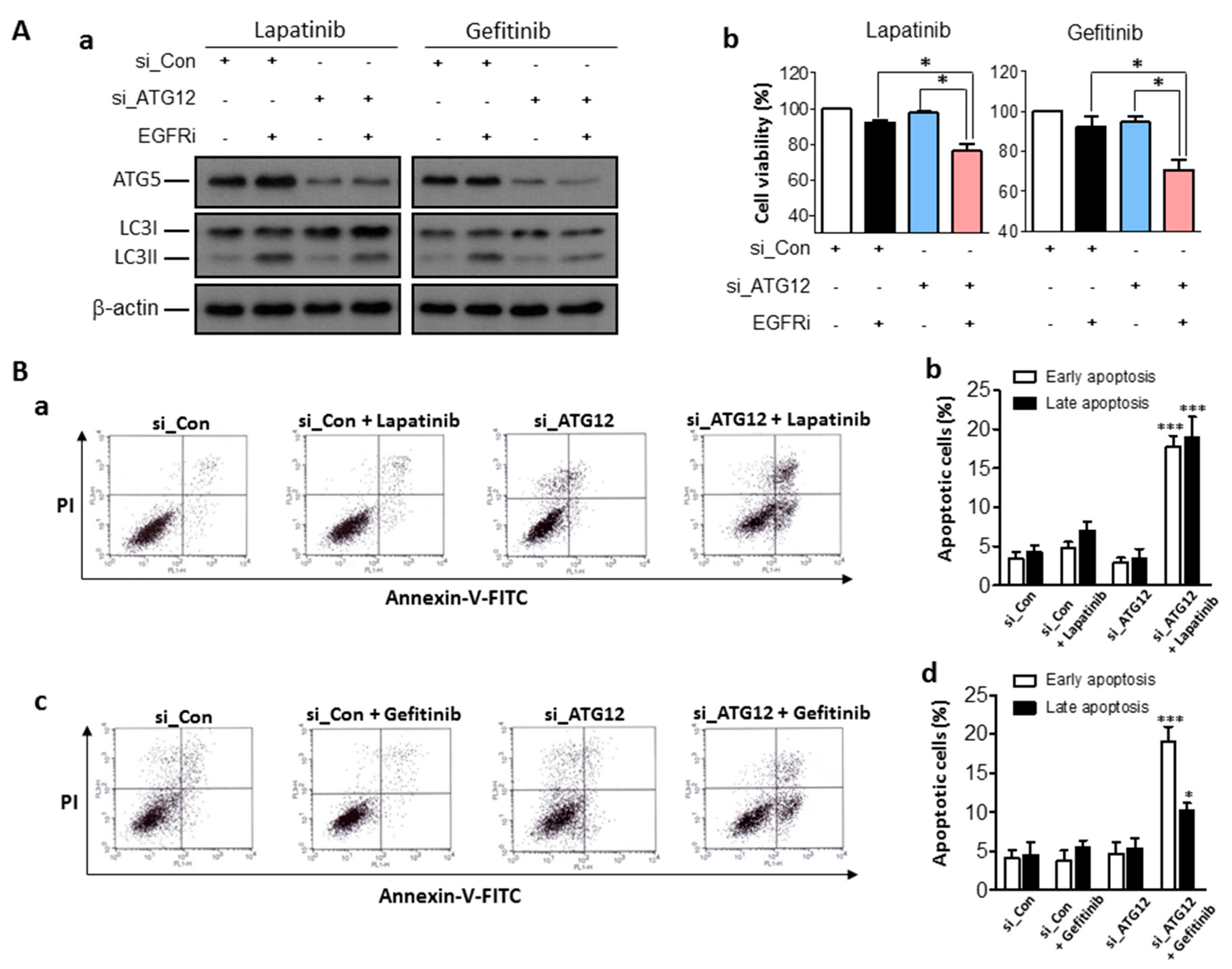
2.4. Enhanced Anti-Cancer Effects of EGFR Inhibitors Combined with Genetic Inhibition of Autophagy Activities in Human Bladder Cancer Cells
3. Discussion
4. Experimental Section
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. Quantitative Real-Time PCR Analysis
4.4. Western Blot Analysis
4.5. Flow Cytometry Analysis of Apoptosis
4.6. Colony Formation Assay
4.7. Generation of LC3-GFP-Expressing T24 Cells and Monitoring of Autophagy Activation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Milowsky, M.I.; Rumble, R.B.; Booth, C.M.; Gilligan, T.; Eapen, L.J.; Hauke, R.J.; Boumansour, P.; Lee, C.T. Guideline on muscle-invasive and metastatic bladder cancer (European Association of Urology Guideline): American society of clinical oncology clinical practice guideline endorsement. J. Clin. Oncol. 2016, 34, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Comperat, E.; Cowan, N.C.; de Santis, M.; Gakis, G.; Lebret, T.; Ribal, M.J.; van der Heijden, A.G.; Sherif, A.; European Association of Urology. EAU guidelines on muscle-invasive and metastatic bladder cancer: Summary of the 2013 guidelines. Eur. Urol. 2014, 65, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Knollman, H.; Godwin, J.L.; Jain, R.; Wong, Y.N.; Plimack, E.R.; Geynisman, D.M. Muscle-invasive urothelial bladder cancer: An update on systemic therapy. Ther. Adv. Urol. 2015, 7, 312–330. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Mooso, B.A.; Vinall, R.L.; Mudryj, M.; Yap, S.A.; deVere White, R.W.; Ghosh, R.M. The role of EGFR family inhibitors in muscle invasive bladder cancer: A review of clinical data and molecular evidence. J. Urol. 2015, 193, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Hu, Y.F.; Feng, X.M.; Tian, T.; Guo, Y.H.; Ma, J.W.; Nan, K.J.; Zhang, H.Y. EGFR inhibitors and autophagy in cancer treatment. Tumour Biol. 2014, 35, 11701–11709. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Rotterud, R.; Nesland, J.M.; Berner, A.; Fossa, S.D. Expression of the epidermal growth factor receptor family in normal and malignant urothelium. BJU Int. 2005, 95, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Kimchi, A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 2009, 14, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Aveic, S.; Tonini, G.P. Resistance to receptor tyrosine kinase inhibitors in solid tumors: Can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell. Int. 2016, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef] [PubMed]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Ito, K.; Yamashiro, Y.; Moriya, S.; Che, X.F.; Yokoyama, T.; Hiramoto, M.; Miyazawa, K. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.C.; Wu, M.Y.; Hwang, M.H.; Chang, Y.T.; Huang, H.J.; Lin, A.M.; Yang, J.C. Chloroquine enhances gefitinib cytotoxicity in gefitinib-resistant nonsmall cell lung cancer cells. PLoS ONE 2015, 10, e0119135. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Lam, S.K.; Mak, J.C.; Zheng, C.Y.; Ho, J.C. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Dilution Factor | Industry |
|---|---|---|---|
| LC3 (microtubule-associated protein light chain 3) | Rabbit | 1:4000 | Novus Biologicals |
| β-Actin | Rabbit | 1:10,000 | Sigma-Aldrich Corporation |
| ATG5 (Autophagy protein 5) | Rabbit | 1:1000 | Cell Signaling Technology |
| p-EGFR (phosphorylated-epidermal growth factor receptor) | Rabbit | 1:1000 | Santa Cruz Biotechnology |
| EGFR (epidermal growth factor receptor) | Rabbit | 1:2000 | Santa Cruz Biotechnology |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.; Lee, K.-H.; Lee, H.S.; Jeong, C.W.; Kwak, C.; Kim, H.H.; Ku, J.H. Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 321. https://doi.org/10.3390/ijms18020321
Kang M, Lee K-H, Lee HS, Jeong CW, Kwak C, Kim HH, Ku JH. Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells. International Journal of Molecular Sciences. 2017; 18(2):321. https://doi.org/10.3390/ijms18020321
Chicago/Turabian StyleKang, Minyong, Kyoung-Hwa Lee, Hye Sun Lee, Chang Wook Jeong, Cheol Kwak, Hyeon Hoe Kim, and Ja Hyeon Ku. 2017. "Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells" International Journal of Molecular Sciences 18, no. 2: 321. https://doi.org/10.3390/ijms18020321
APA StyleKang, M., Lee, K. -H., Lee, H. S., Jeong, C. W., Kwak, C., Kim, H. H., & Ku, J. H. (2017). Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells. International Journal of Molecular Sciences, 18(2), 321. https://doi.org/10.3390/ijms18020321