
Effects of the Activin A–Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure

Abstract
:1. Introduction
2. Results
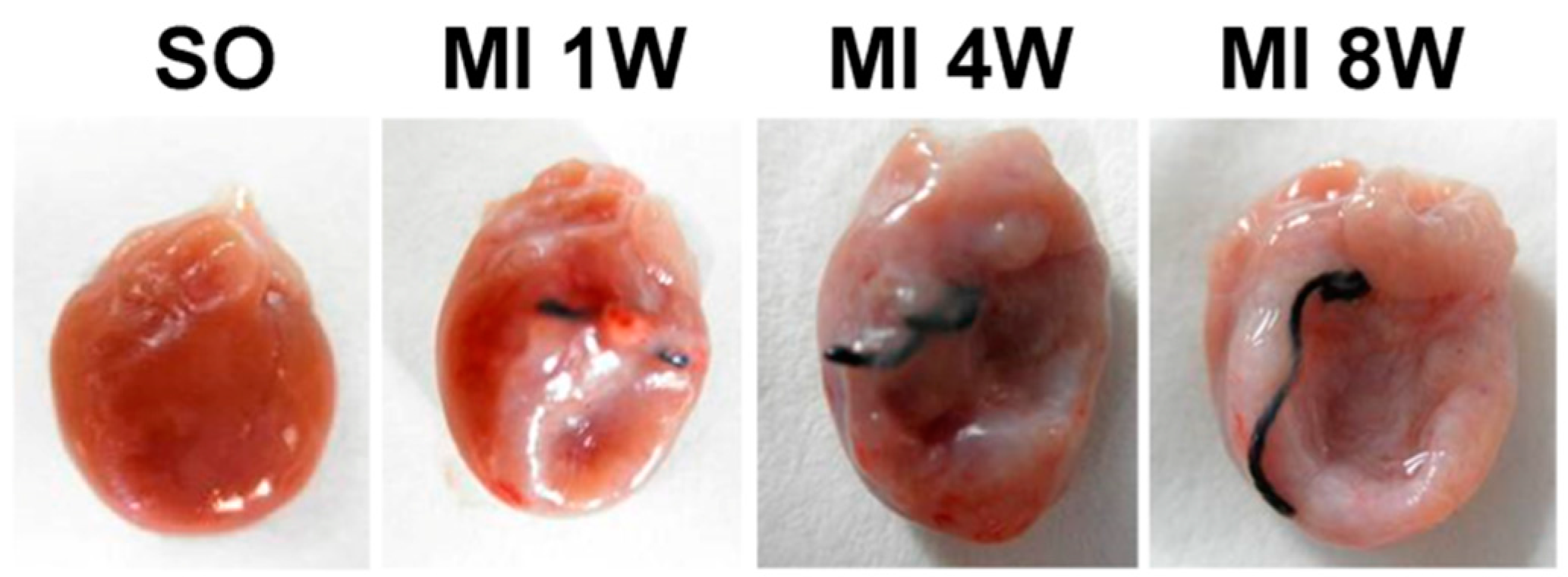
2.1. General Observations
2.2. Morphological and Hemodynamic Characteristics of Rats
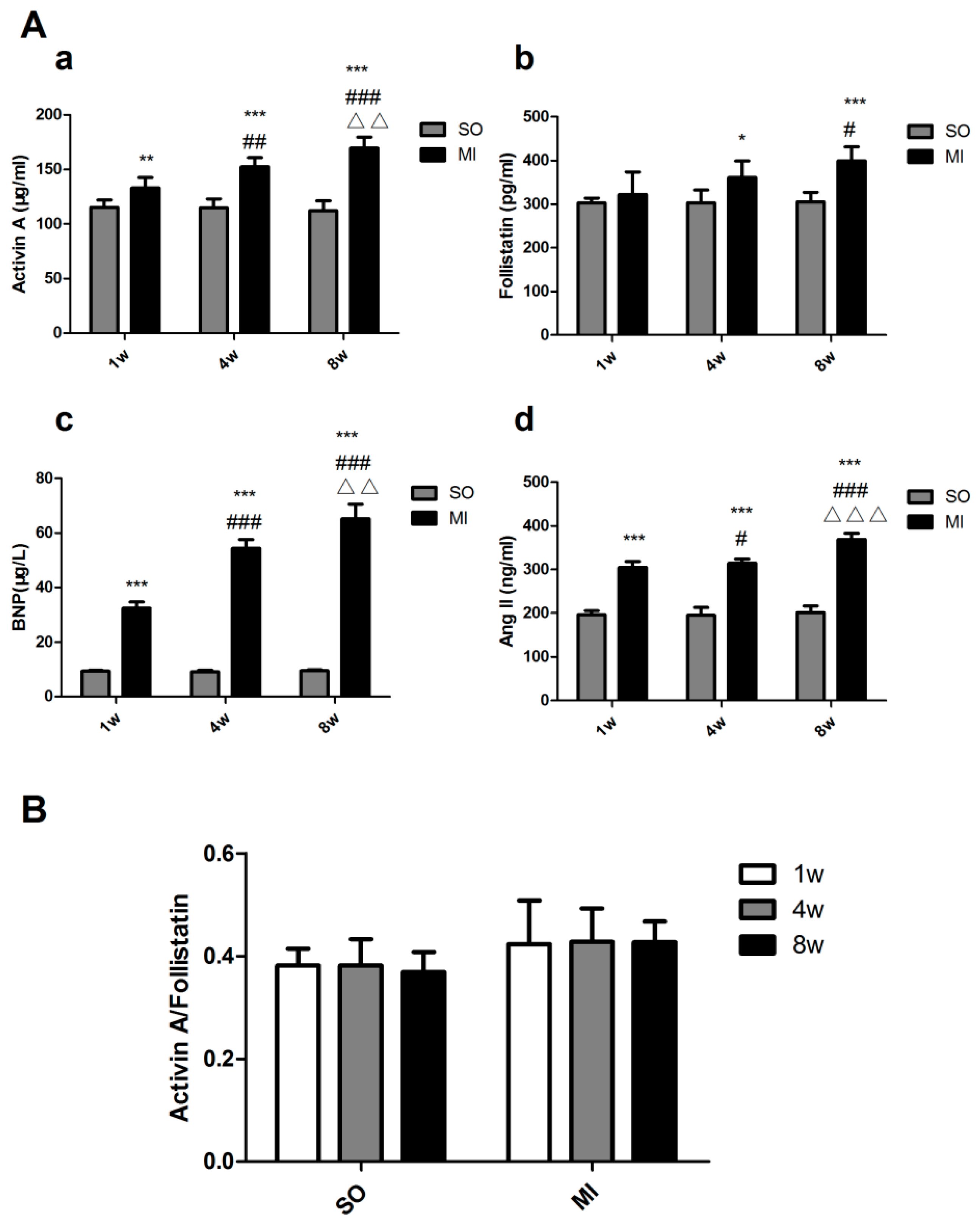
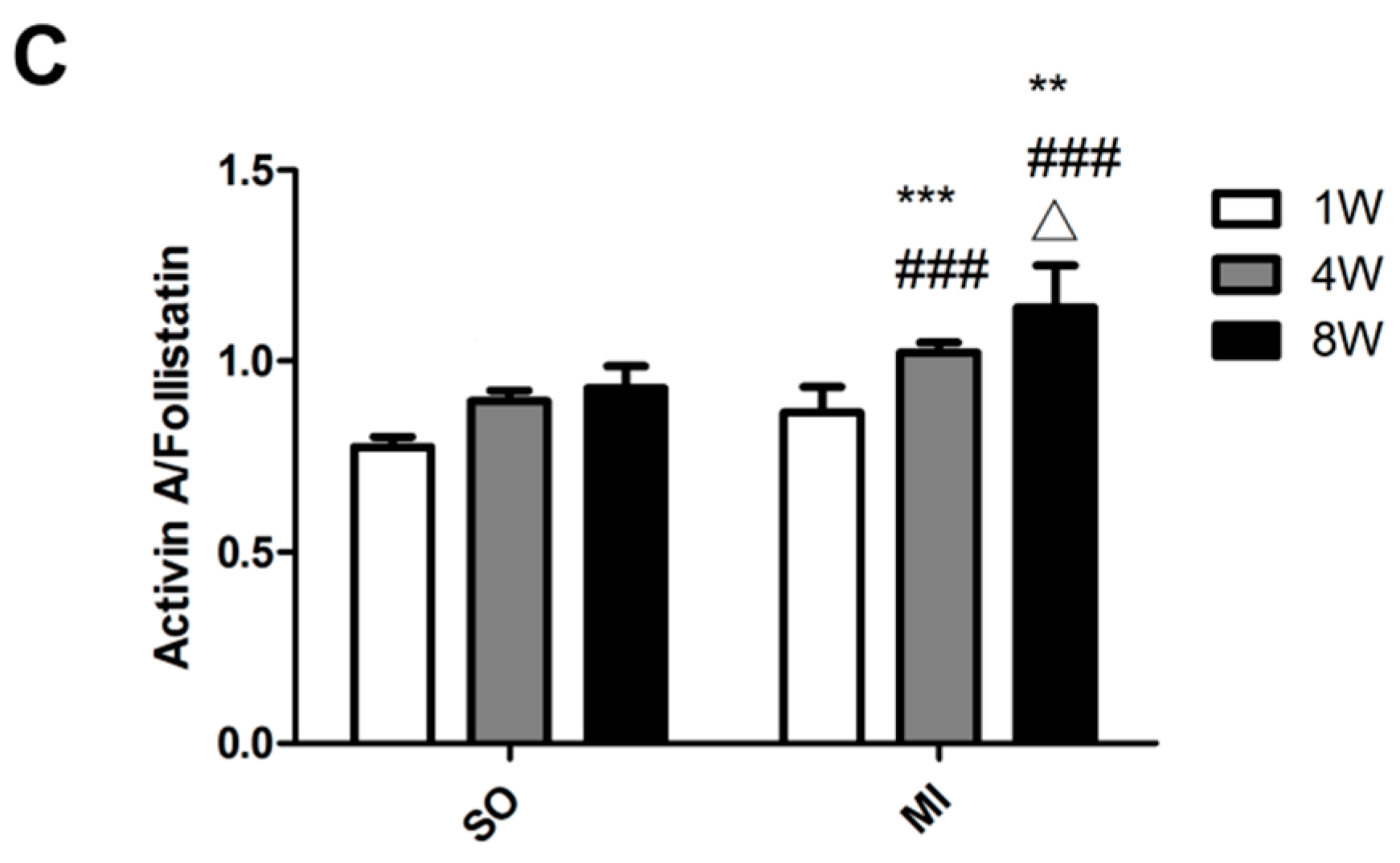
2.3. Expression Levels of Activin A–Follistatin System and Brain Natriuretic Peptide (BNP), Angiotensin II (Ang II) in the Serum of Rats
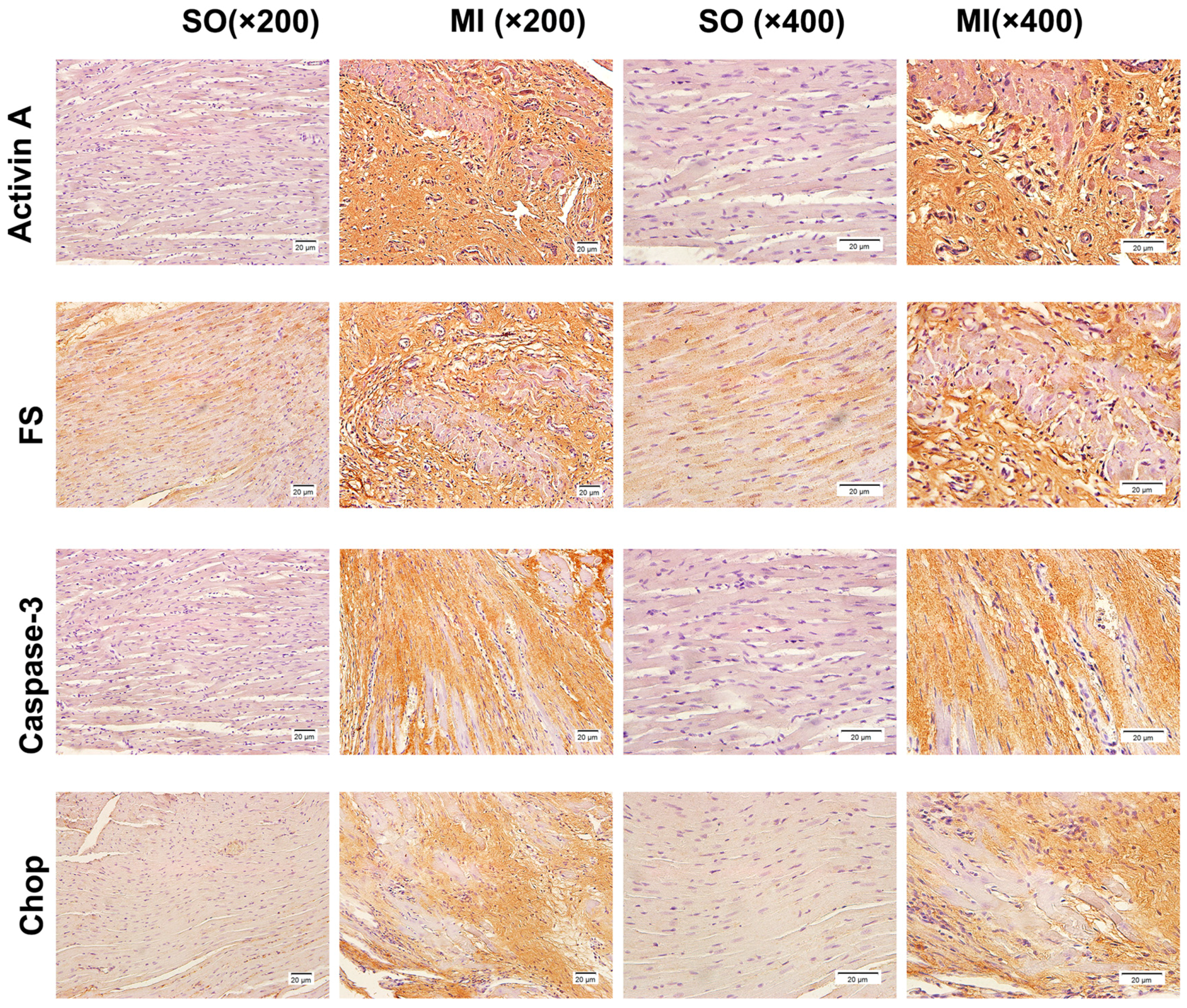
2.4. Detection of the Expression of Activin A, Follistatin, Caspase-3, and C/EBP Homologous Protein (CHOP) by Immunohistochemical Staining
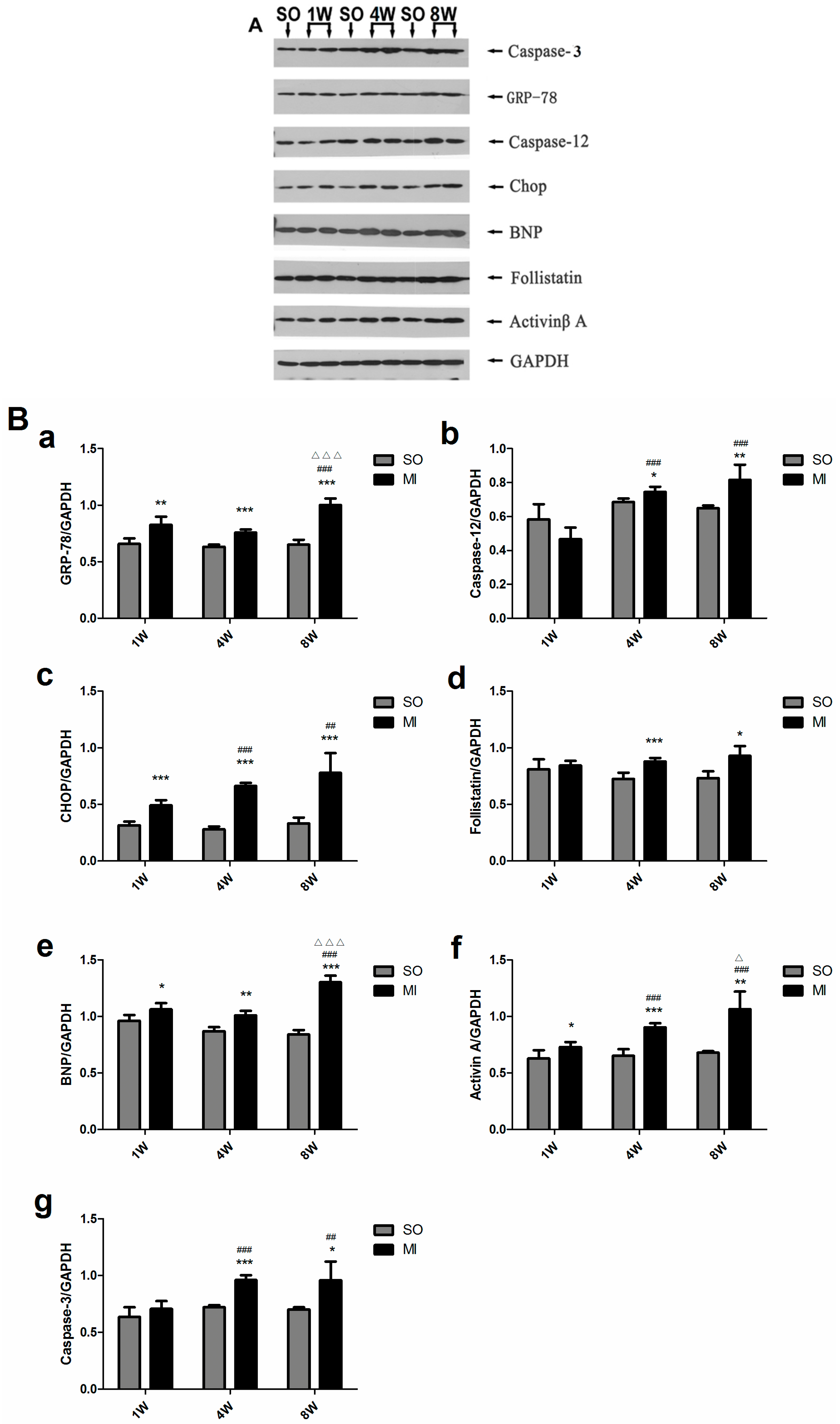
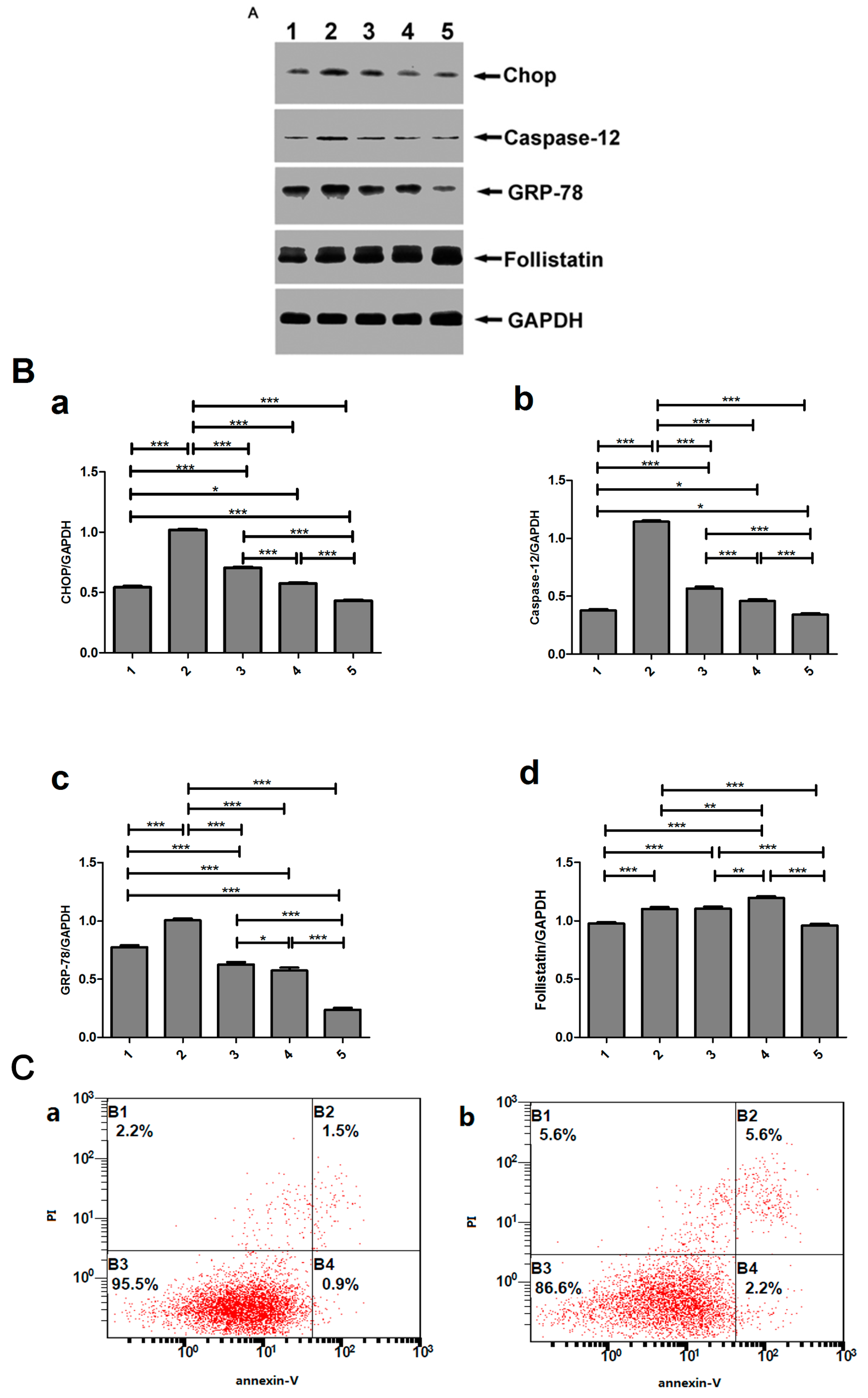
2.5. Detection of the Expression of Activin A, Follistatin, BNP, Caspase-3 and Endoplasmic Reticulum Stress (ERS)-Related Molecules in the Left Ventricular Noninfarction Area by Western Blot
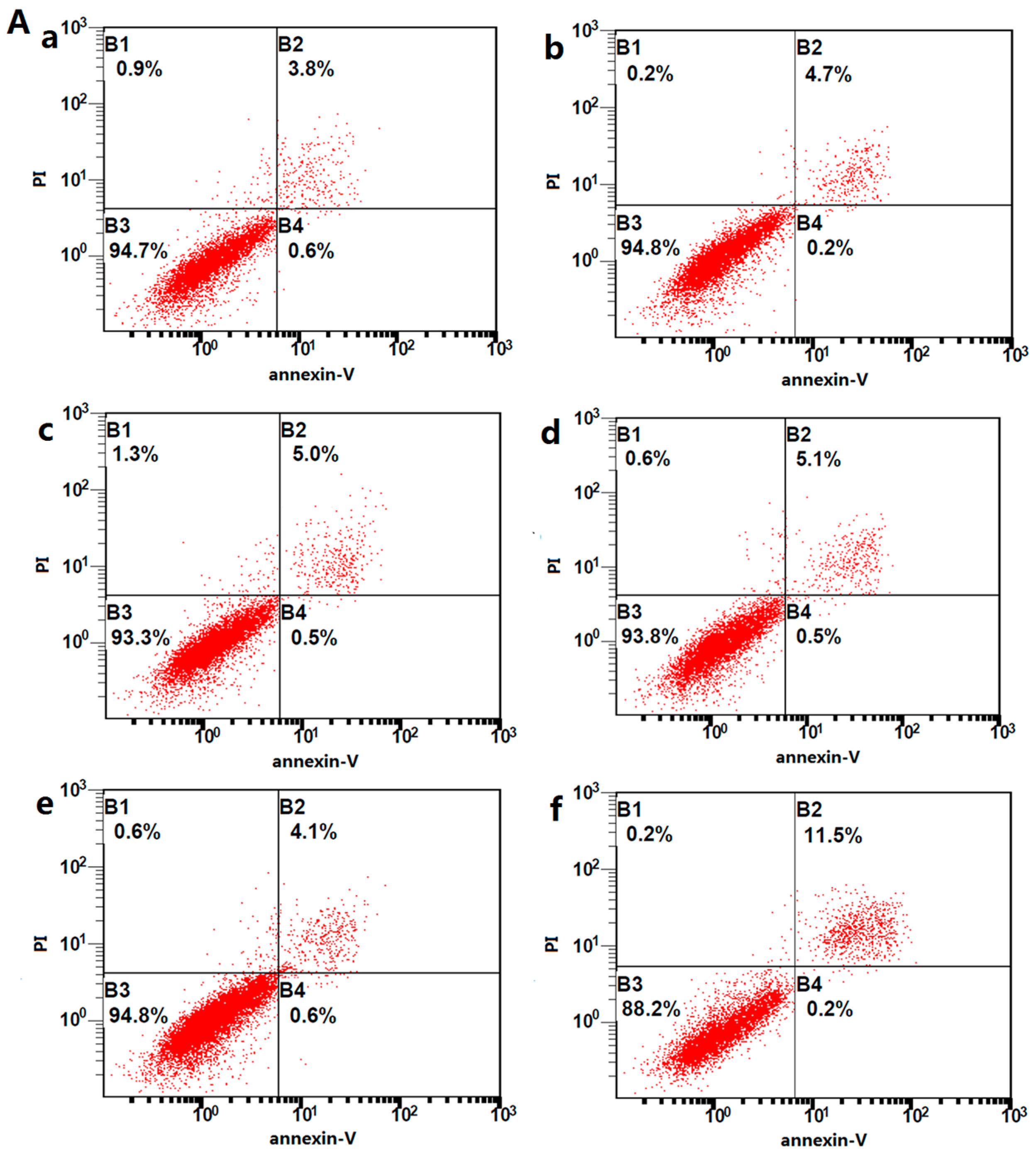
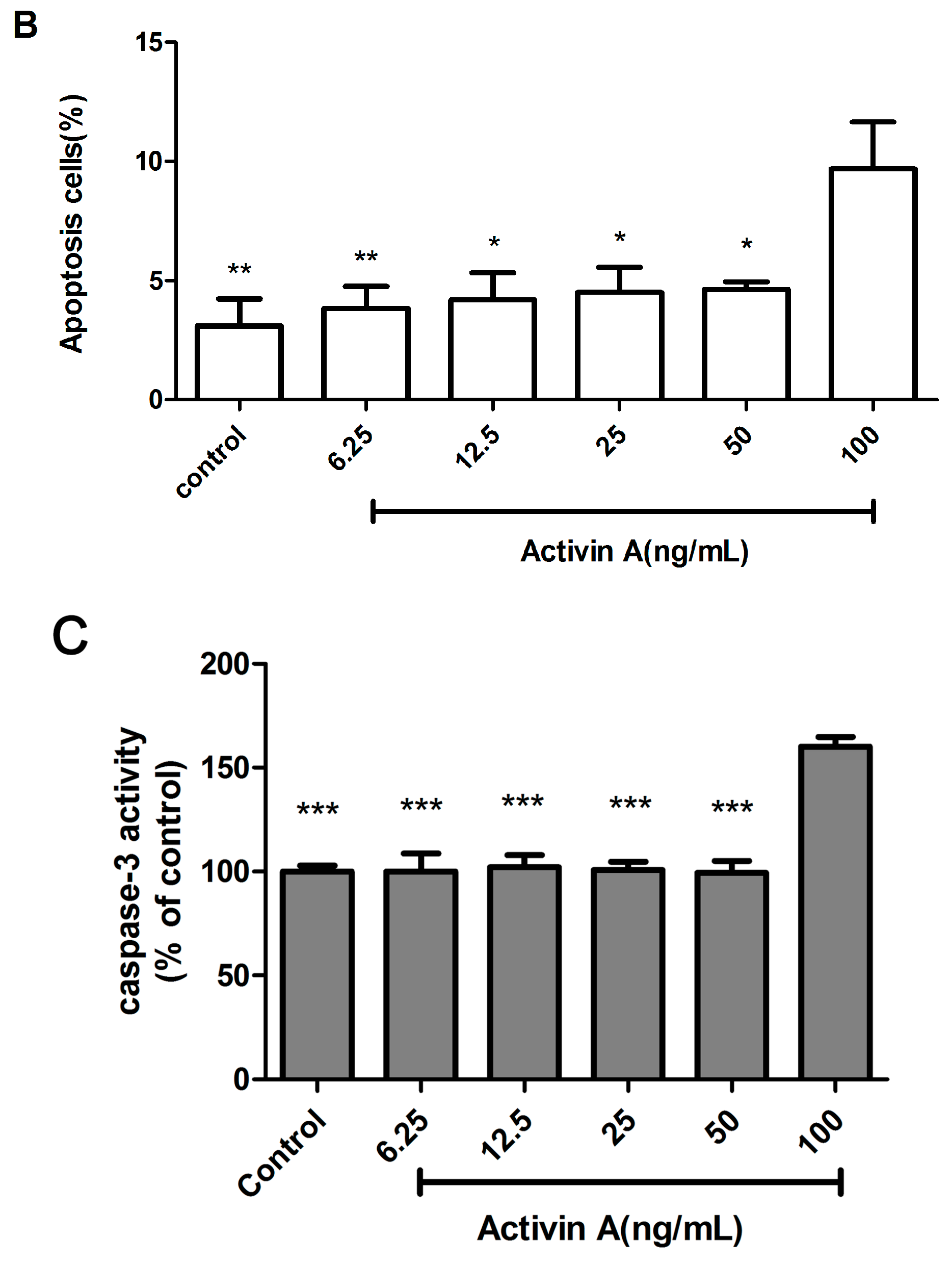
2.6. Effect of Different Concentrations of Activin A on H9c2 Cell Apoptosis
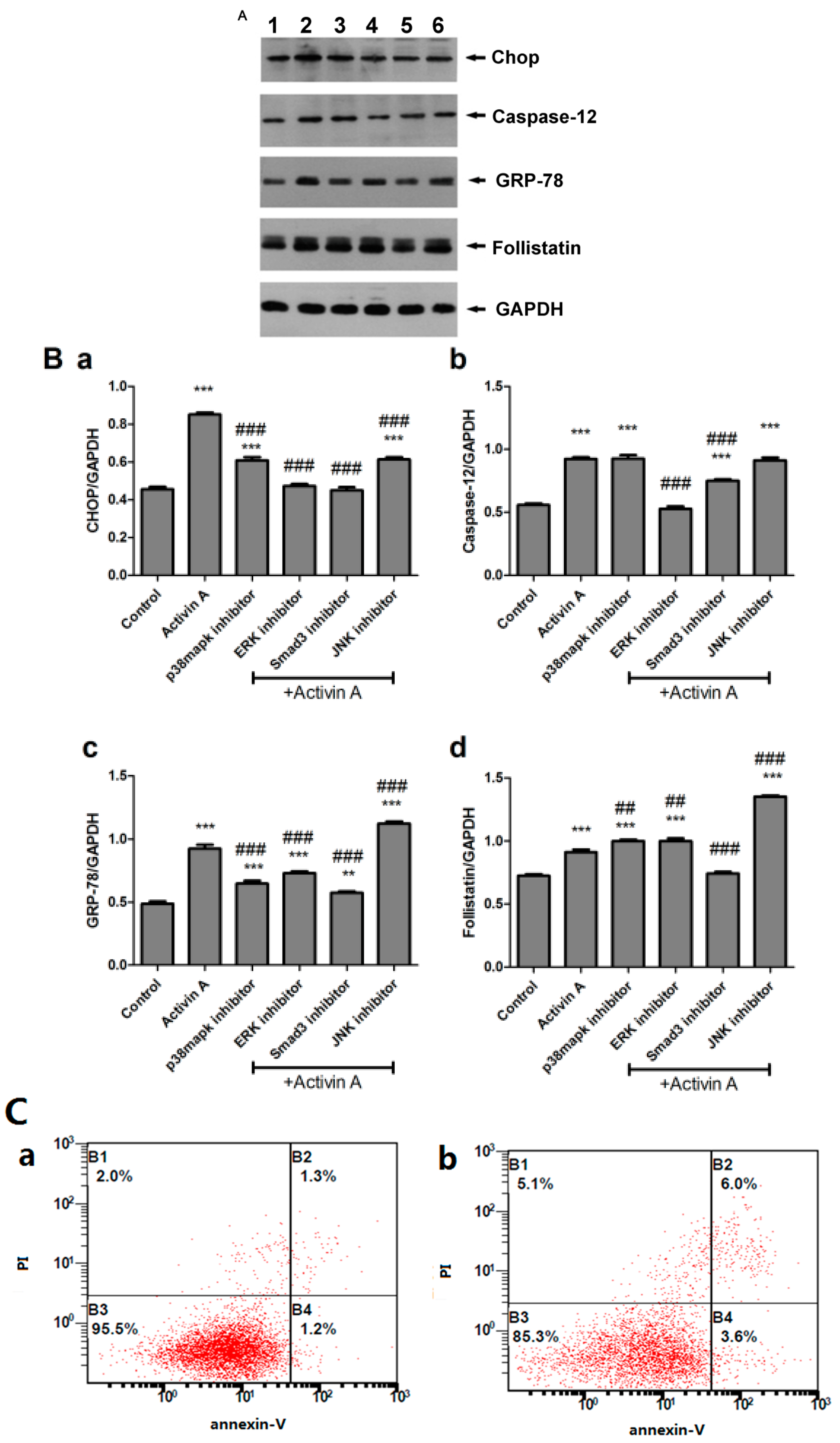
2.7. Use of High Concentration (100 ng/mL) of Activin A to Show that the Activin A–Follistatin System Regulated ERS-Mediated Cardiomyocyte Apoptosis In Vitro
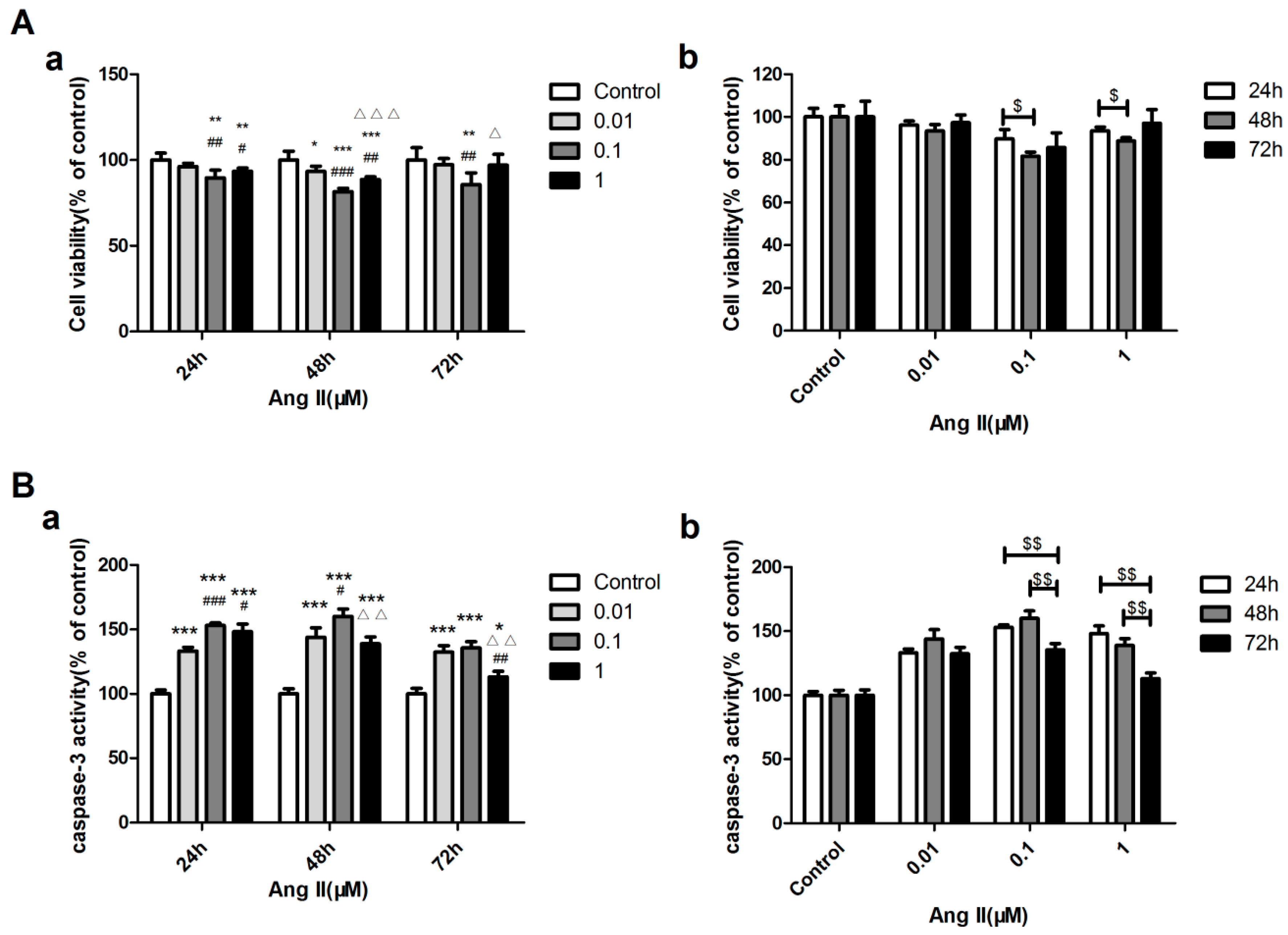
2.8. Concentration and Acting Time of Ang II-Stimulated H9c2 Cell Apoptosis
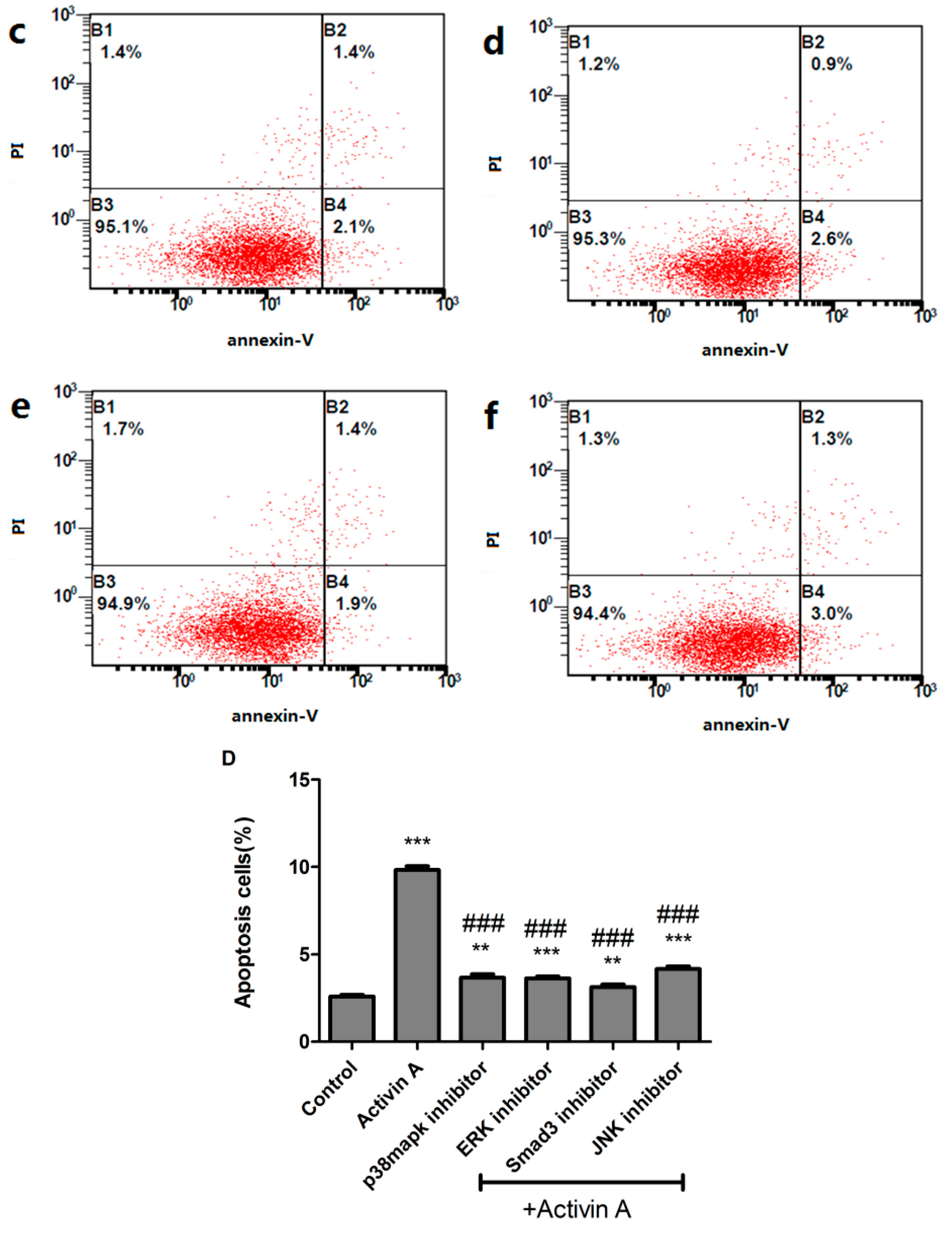
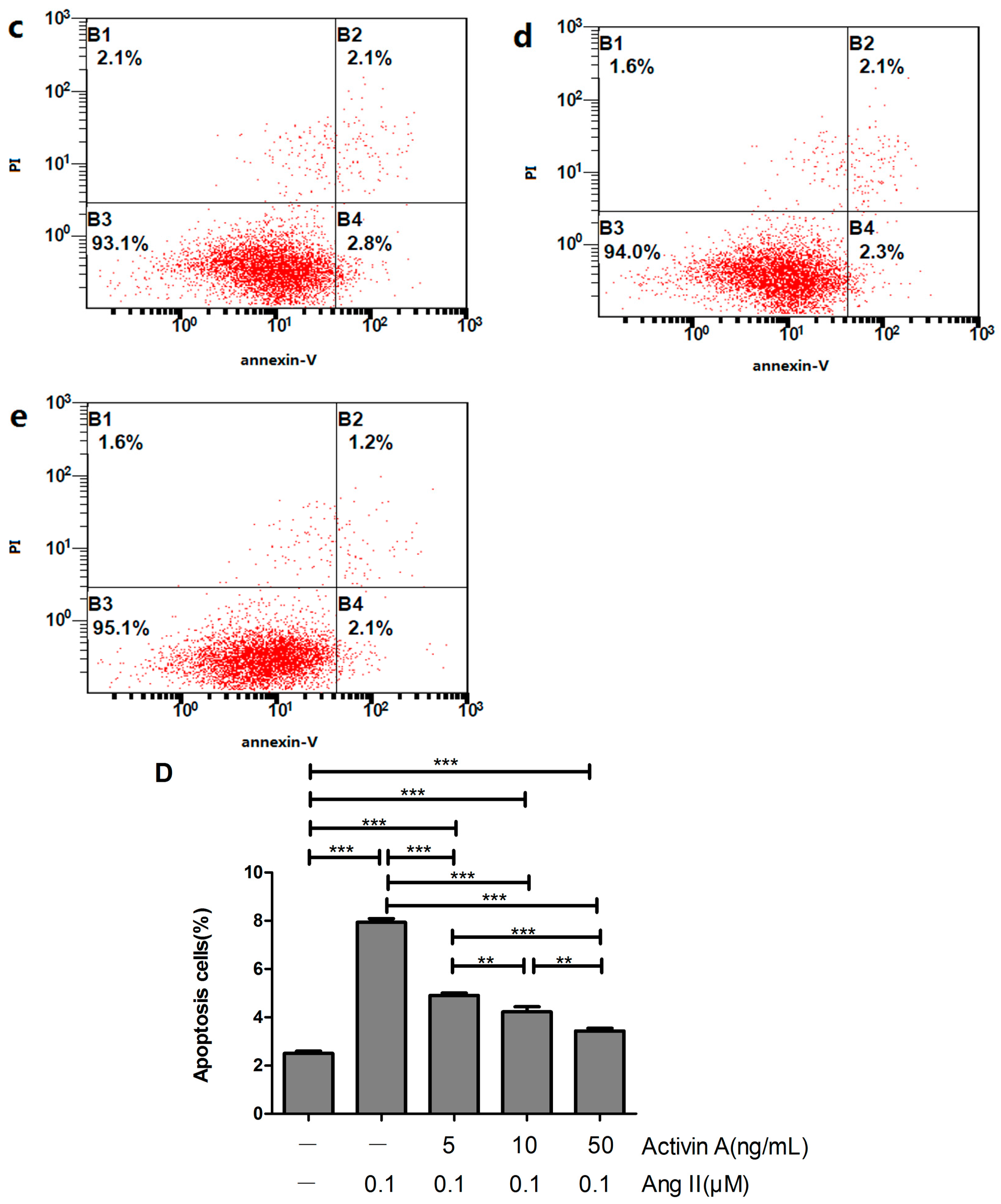
2.9. Effect of Low Concentration of Activin A on Ang II-Induced Cardiomyocyte Apoptosis and Its Relationship with ERS
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Establishment of the Rat Model with Heart Failure (HF)
4.3. Hemodynamic Investigation
4.4. ELISA
4.5. Immunohistochemical Staining
4.6. Cell Culture
4.7. MTT Assay
4.8. Flow Cytometry
4.9. Assay of Caspase-3 Activity
4.10. Western Blot
4.11. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Werner, S.; Alzheimer, C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006, 17, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.Y.; Zhang, Z.; Furst, B.; Batres, Y.; Huang, G.; Li, G. Activins and activin receptors in cell growth. Proc. Soc. Exp. Biol. Med. 1997, 214, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hubner, G.; Alzheimer, C.; Werner, S. Activin: A novel player in tissue repair processes. Histol. Histopathol. 1999, 14, 295–304. [Google Scholar] [PubMed]
- Hayashi, Y.; Maeshima, K.; Goto, F.; Kojima, I. Activin A as a critical mediator of capillary formation: Interaction with the fibroblast growth factor action. Endocr. J. 2007, 54, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Munz, B.; Smola, H.; Engelhardt, F.; Bleuel, K.; Brauchle, M.; Lein, I.; Evans, L.W.; Huylebroeck, D.; Balling, R.; Werner, S. Overexpression of activin A in the skin of transgenic mice reveals new activities of activin in epidermal morphogenesis, dermal fibrosis and wound repair. EMBO J. 1999, 18, 5205–5215. [Google Scholar] [CrossRef] [PubMed]
- Aoki, F.; Kurabayashi, M.; Hasegawa, Y.; Kojima, I. Attenuation of bleomycin-induced pulmonary fibrosis by follistatin. Am. J. Respir. Crit. Care Med. 2005, 172, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Matsuse, T.; Ikegami, A.; Ohga, E.; Hosoi, T.; Oka, T.; Kida, K.; Fukayama, M.; Inoue, S.; Nagase, T.; Ouchi, Y.; Fukuchi, Y. Expression of immunoreactive activin A protein in remodeling lesions associated with interstitial pulmonary fibrosis. Am. J. Pathol. 1996, 148, 707–713. [Google Scholar] [PubMed]
- Ohga, E.; Matsuse, T.; Teramoto, S.; Katayama, H.; Nagase, T.; Fukuchi, Y.; Ouchi, Y. Effects of activin A on proliferation and differentiation of human lung fibroblasts. Biochem. Biophys. Res. Commun. 1996, 228, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Ohga, E.; Matsuse, T.; Teramoto, S.; Ouchi, Y. Activin receptors are expressed on human lung fibroblast and activin A facilitates fibroblast-mediated collagen gel contraction. Life Sci. 2000, 66, 1603–1613. [Google Scholar] [CrossRef]
- Wada, W.; Kuwano, H.; Hasegawa, Y.; Kojima, I. The dependence of transforming growth factor-β-induced collagen production on autocrine factor activin A in hepatic stellate cells. Endocrinology 2004, 145, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.J.; Francis, R.J.; Zimmermann, A.; Mellor, SL.; Cranfield, M.; Risbridger, G.P.; Groome, N.P.; Wheatley, A.M.; Fleming, J.S. Changes in activin and activin receptor subunit expression in rat liver during the development of CCl4-induced cirrhosis. Mol. Cell. Endocrinol. 2003, 201, 143–153. [Google Scholar] [CrossRef]
- Ohnishi, N.; Miyata, T.; Ohnishi, H.; Yasuda, H.; Tamada, K.; Ueda, N.; Mashima, H.; Sugano, K. Activin A is an autocrine activator of rat pancreatic stellate cells: Potential therapeutic role of follistatin for pancreatic fibrosis. Gut 2003, 52, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Maeshima, A.; Kojima, I.; Nojima, Y. Activin A is a potent activator of renal interstitial fibroblasts. J. Am. Soc. Nephrol. 2004, 15, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Gaedeke, J.; Boehler, T.; Budde, K.; Neumayer, H.H.; Peters, H. Glomerular activin A overexpression is linked to fibrosis in anti-Thy1 glomerulonephritis. Nephrol. Dial. Transplant. 2005, 20, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Lahme, B.; Siluschek, M.; Rehbein, K.; Weiskirchen, R.; Gressner, A.M. Intracrine signalling of activin A in hepatocytes upregulates connective tissue growth factor (CTGF/CCN2) expression. Liver Int. 2008, 28, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Y.N.; Cui, X.L.; Fang, S.Y.; Ge, J.Y.; Sun, Y.; Liu, Z.H. The role and mechanism of action of activin A in neurite outgrowth of chicken embryonic dorsal root ganglia. J. Cell Sci. 2012, 125, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Wankell, M.; Munz, B.; Hübner, G.; Hans, W.; Wolf, E.; Goppelt, A.; Werner, S. Impaired wound healing in transgenic mice overexpressing the activin antagonist follistatin in the epidermis. EMBO J. 2001, 20, 5361–5372. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Mine, T.; Shibata, H.; Eto, Y.; Hasegawa, Y.; Takeuchi, T.; Asano, S.; Kojima, I. Activin A: An autocrine inhibitor of initiation of DNA synthesis in rat hepatocytes. J. Clin. Investig. 1993, 92, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Schwall, R.H.; Robbins, K.; Jardieu, P.; Chang, L.; Lai, C.; Terrell, T.G. Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology 1993, 18, 347–356. [Google Scholar] [CrossRef]
- Hully, J.R.; Chang, L.; Schwall, R.H.; Widmer, H.R.; Terrell, T.G.; Gillett, N.A. Induction of apoptosis in the murine liver with recombinant human activin A. Hepatology 1994, 20, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Ouchi, N.; Shimano, M.; Pimentel, D.R.; Papanicolaou, K.N.; Panse, K.D.; Tsuchida, K.; Lara-Pezzi, E.; Lee, S.J.; Walsh, K. Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation 2009, 120, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, S.; Kirshner, A.; de Moissac, D.; Kirshenbaum, L.A. A direct requirement of nuclear factor-κB for suppression of apoptosis in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 939–945. [Google Scholar]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Kimes, B.W.; Brandt, B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Ekhterae, D.; Lin, Z.; Lundberg, M.S.; Crow, M.T.; Brosius, F.C.; Núñez, G. ARC inhibits cytochrome c release from mitochondria and protects against hypoxia-induced apoptosis in heart-derived H9c2 cells. Circ. Res. 1999, 85, 70–77. [Google Scholar] [CrossRef]
- Unger, T.; Li, J. The role of the renin-angiotensin-aldosterone system in heart failure. J. Renin. Angiotensin Aldosterone Syst. 2004, 5, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Ainscough, J.F.; Drinkhill, M.J.; Sedo, A.; Sedo, A.; Turner, N.A.; Brooke, D.A.; Balmforth, A.J.; Ball, S.G. Angiotensin-II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc. Res. 2009, 81, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Mel’nikova, N.P.; Timoshin, S.S.; Jivotova, E.Y.; Pelliniemi, L.J.; Jokinen, E.; Abdelwahid, E. Angiotensin-II activates apoptosis, proliferation and protein synthesis in the left heart ventricle of newborn albino rats. Int. J. Cardiol. 2006, 112, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, G.; Wang, Y.; Liu, Q.; Xu, W.; Tan, Y.; Cai, L. Diabetes- and angiotensin-II-induced cardiac endoplasmic reticulum stress and cell death: Metallothionein protection. J. Cell. Mol. Med. 2009, 13, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Westphal, E.; Buchhorn, R.; Wessel, A.; Werdan, K. Interleukin-1 and related proteins in cardiovascular disease in adults and children. Shock 2001, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Takimoto, Y.; Pennica, D.; Inoue, R.; Shinoda, E.; Hattori, R.; Yui, Y.; Sasayama, S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J. Mol. Cell. Cardiol. 2000, 32, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Pollock, D.M.; Pollock, J.S. Endothelin and oxidative stress in the vascular system. Curr. Vasc. Pharmacol. 2005, 3, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-β signaling through the Smad pathway: Role in extracellular matrix gene expression and regulation. J. Investig. Dermatol. 2002, 118, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudabady, M.; Mathieu, M.; Dewachter, L.; Hadad, I.; Ray, L.; Jespers, P.; Brimioulle, S.; Naeije, R.; McEntee, K. Activin-A, transforming growth factor-β, and myostatin signaling pathway in experimental dilated cardiomyopathy. J. Card. Fail. 2008, 14, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Ueland, T.; Øie, E.; Florholmen, G.; Halvorsen, B.; Attramadal, H.; Simonsen, S.; Frøland, S.S.; Gullestad, L.; Christensen, G.; et al. Elevated levels of activin A in heart failure: Potential role in myocardial remodeling. Circulation 2004, 109, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Wang, Y.N.; Liu, H.Y.; Yang, J.; Yang, C.Y.; Liu, M.; Liu, Y.F.; Yang, P.; Liu, Z.H. The expression and role of activin A and follistatin in heart failure rats after myocardial infarction. Int. J. Cardiol. 2013, 168, 2994–2997. [Google Scholar] [CrossRef] [PubMed]
- Panse, K.D.; Felkin, L.E.; López-Olañeta, M.M.; Gómez-Salinero, J.; Villalba, M.; Muñoz, L.; Nakamura, K.; Shimano, M.; Walsh, K.; Barton, P.J.; et al. Follistatin-like 3 mediates paracrine fibroblast activation by cardiomyocytes. J. Cardiovasc. Transl. Res. 2012, 5, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Shimano, M.; Ouchi, N.; Nakamura, K.; Oshima, Y.; Higuchi, A.; Pimentel, D.R.; Panse, K.D.; Lara-Pezzi, E.; Lee, S.J.; Sam, F.; et al. Cardiac myocyte-specific ablation of follistatin-like 3 attenuates stress-induced myocardial hypertrophy. J. Biol. Chem. 2011, 286, 9840–9848. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Ren, J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: Role of insulin signaling and ER stress. J. Mol. Cell. Cardiol. 2008, 44, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Iwai, C.; Liu, J.; Sheu, S.S.; Fu, M.; Liang, C.S. Darbepoetin alfa exerts a cardioprotective effect in autoimmune cardiomyopathy via reduction of ER stress and activation of the PI3K/Akt and STAT3 pathways. J. Mol. Cell. Cardiol. 2008, 45, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Q.; Yang, C.Y.; Li, S.M.; Liu, M.; Ding, M.; Liu, G.H.; Yang, P. Lipopolysaccharide induced activin A–follistatin imbalance affects cardiac fibrosis. Chin. Med. J. 2012, 125, 2205–2212. [Google Scholar] [PubMed]
- Yang, C.; Wang, Y.; Liu, H.; Li, N.; Sun, Y.; Liu, Z.; Yang, P. Ghrelin protects H9c2 cardiomyocytes from angiotensin II-induced apoptosis through the endoplasmic reticulum stress pathway. J. Cardiovasc. Pharmacol. 2012, 59, 465–471. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size of Heart (cm) | SO | MI | ||||
|---|---|---|---|---|---|---|
| 1 W | 4 W | 8 W | 1 W | 4 W | 8 W | |
| long diameter | 1.383 ± 0.183 | 1.483 ± 0.075 | 1.533 ± 0.081 | 1.4 ± 0.19 | 1.65 ± 0.055 **,## | 1.75 ± 0.084 **,##,Δ |
| short diameter | 0.15 ± 0.105 | 1.233 ± 0.052 | 1.217 ± 0.117 | 1.067 ± 0.121 | 1.3 ± 0.167 # | 1.367 ± 0.121 ## |
| Characteristics | SO | MI | ||||
|---|---|---|---|---|---|---|
| 1 W | 4 W | 8 W | 1 W | 4 W | 8 W | |
| HW/BW (mg/g) | 3.20 ± 0.098 | 3.21 ± 0.130 | 3.19 ± 0.163 | 3.86 ± 0.217 *** | 3.80 ± 0.219 *** | 3.52 ± 0.158 **,#,Δ |
| LVW/BW (mg/g) | 2.24 ± 0.130 | 2.22 ± 0.145 | 2.18 ± 0.192 | 2.68 ± 0.105 *** | 2.50 ± 0.206 * | 2.46 ± 0.155 *,# |
| HR (/min) | 435 ± 31.45 | 437.33 ± 31.10 | 430 ± 52.31 | 413.83 ± 50.70 | 410.66 ± 68.24 | 353.67 ± 37.47 *,# |
| SBP (mmHg) | 98.97 ± 12.67 | 98.89 ± 15.13 | 97.03 ± 13.06 | 89.79 ± 14.01 | 81.84 ± 19.14 | 63.78 ± 5.61 ***,## |
| DBP (mmHg) | 59.96 ± 3.67 | 76.34 ± 17.73 | 66.79 ± 10.87 | 75.22 ± 15.72 * | 69.97 ± 20.59 | 53.06 ± 5.19 *,## |
| LVSP (mmHg) | 115.98 ± 3.53 | 110.48 ± 18.51 | 105.15 ± 12.19 | 63.12 ± 8.96 *** | 60.06 ± 3.96 *** | 47.90 ± 7.50 ***,##,ΔΔ |
| LVEDP (mmHg) | 3.82 ± 9.28 | 3.745 ± 10.04 | 3.665 ± 9.84 | 22.76 ± 7.19 ** | 23.97 ± 8.35 ** | 24.15 ± 4.83 ** |
| +dp/dtmax (mmHg/s) | 5455 ± 443.92 | 5373.33 ± 842.29 | 5465 ± 974.77 | 2720 ± 248.83 *** | 2498.33 ± 251.42 *** | 2070 ± 140.28 ***,###,ΔΔ |
| −dp/dtmax (mmHg/s) | 3885 ± 321.66 | 4460 ± 989.86 | 4146.667 ± 817.32 | 2171.667 ± 216.92 *** | 2171.667 ± 172.55 *** | 1850 ± 235.96 ***,#,Δ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Mao, C.; Li, J.; Han, F.; Yang, P. Effects of the Activin A–Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure. Int. J. Mol. Sci. 2017, 18, 374. https://doi.org/10.3390/ijms18020374
Liu M, Mao C, Li J, Han F, Yang P. Effects of the Activin A–Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure. International Journal of Molecular Sciences. 2017; 18(2):374. https://doi.org/10.3390/ijms18020374
Chicago/Turabian StyleLiu, Miao, Cuiying Mao, Jiayu Li, Fanglei Han, and Ping Yang. 2017. "Effects of the Activin A–Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure" International Journal of Molecular Sciences 18, no. 2: 374. https://doi.org/10.3390/ijms18020374
APA StyleLiu, M., Mao, C., Li, J., Han, F., & Yang, P. (2017). Effects of the Activin A–Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure. International Journal of Molecular Sciences, 18(2), 374. https://doi.org/10.3390/ijms18020374