
Clarifying the Ghrelin System’s Ability to Regulate Feeding Behaviours Despite Enigmatic Spatial Separation of the GHSR and Its Endogenous Ligand

Abstract
:1. Introduction
1.1. Preface
1.2. Ghrelin: The Feeding Peptide
1.3. Ghrelin’s Hypothalamic Modulation of Feeding
1.4. Ghrelin’s Hedonic Regulation of Feeding
2. The Ghrelin and Growth Hormone Secretagogue Receptor Paradox
2.1. Peripheral Ghrelin Activates Central Targets
2.2. The Blood Brain Barrier
2.3. Permeability of the Blood Brain Barrier to Ghrelin
3. Peripheral Ghrelin Stimulates Appetite
3.1. Vagal Afferent Activation by Ghrelin
3.2. Ghrelin and Circumventricular Organs
3.2.1. Median Eminence
3.2.2. Subfornical Organ
3.2.3. Area Postrema
4. The Role of Central Ghrelin in Stimulating Feeding
4.1. Central c-Fos Studies
4.2. Cerebrospinal Fluid Ghrelin
4.3. Central Ghrelin Synthesis
4.3.1. Support for the Central Synthesis of Ghrelin
4.3.2. Evidence Opposing the Central Synthesis of Ghrelin
4.3.3. Future Avenues to Ease the Central Synthesis of Ghrelin Debate
5. GHSR Activity Independent of Ghrelin
5.1. GHSR Constitutive Activity
5.2. GHSR Promiscuity
5.3. GHSR Dimers
5.4. GHSR and Dopamine Receptor Heterodimers
5.5. GHSR/5-Hydroxytryptamine2C Heterodimers
5.6. GHSR/Melanocortin Receptor 3 Heterodimers
6. Conclusions
Conflicts of Interest
References
- Abizaid, A.; Liu, Z.W.; Andrews, Z.B.; Shanabrough, M.; Borok, E.; Elsworth, J.D.; Roth, R.H.; Sleeman, M.W.; Picciotto, M.R.; Tschöp, M.H.; et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Investig. 2006, 116, 3229–3239. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Inui, A.; Kaga, T.; Yuzuriha, H.; Nagata, T.; Fujimiya, M.; Katsuura, G.; Makino, S.; Fujino, M.A.; Kasuga, M. A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology 2001, 74, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Diano, S.; Farr, S.A.; Benoit, S.C.; McNay, E.C.; da Silva, I.; Horvath, B.; Gaskin, F.S.; Nonaka, N.; Jaeger, L.B.; Banks, W.A.; et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 2006, 9, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.D.; Feighner, S.D.; Cully, D.F.; Arena, J.P.; Paul, A.; Rosenblum, C.I.; Hamelin, M.; Hreniuk, D.L.; Palyha, O.C.; Hamelin, M.; et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 1996, 273, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Lutter, M.; Sakata, I.; Osborne-Lawrence, S.; Rovinsky, S.A.; Anderson, J.G.; Jung, S.; Birnbaum, S.; Yanagisawa, M.; Elmquist, J.K.; Nestler, E.J.; et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat. Neurosci. 2008, 11, 752–753. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nature 2001, 409, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Finger, B.C.; Dinan, T.G.; Cryan, J.F. Ghrelin signalling and obesity: At the interface of stress, mood and food reward. Pharmacol. Ther. 2012, 135, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, P.; Zheng, H.; Smith, R.G. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 4679–4684. [Google Scholar] [CrossRef] [PubMed]
- Wren, A.M.; Seal, L.J.; Cohen, M.A.; Brynes, A.E.; Frost, G.S.; Murphy, K.G.; Dhillo, W.S.; Ghatei, M.A.; Bloom, S.R. Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 2001, 86, 5992–5995. [Google Scholar] [CrossRef] [PubMed]
- Wren, A.M.; Small, C.J.; Abbott, C.R.; Shillo, W.S.; Seal, L.J.; Cohen, M.A.; Batterham, R.L.; Taheri, S.; Stanley, S.A.; Ghatei, M.A.; et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes 2001, 50, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; Salio, C.; Lossi, L.; Merighi, A. Ghrelin in central neurons. Curr. Neuropharmacol. 2009, 7, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 2002, 143, 3179–3182. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Yu, H.; Palyha, O.; McKee, K.; Feighner, S.; Sirinathsinghji, D.; Smith, R.; van der Ploeg, L.; Howard, A. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Mol. Brain Res. 1997, 48, 23–29. [Google Scholar] [CrossRef]
- Mani, B.K.; Walker, A.K.; Lopez Soto, E.J.; Raingo, J.; Lee, C.E.; Perelló, M.; Andrews, Z.B.; Zigman, J.M. Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J. Comp. Neurol. 2014, 522, 3644–3666. [Google Scholar] [CrossRef] [PubMed]
- Papotti, M.; Ghè, C.; Cassoni, P.; Catapano, F.; Deghenghi, R.; Ghigo, E.; Muccioli, G. Growth hormone secretagogue binding sites in peripheral human tissues. J. Clin. Endocrinol. Metab. 2000, 85, 3803–3807. [Google Scholar] [CrossRef] [PubMed]
- Shuto, Y.; Shibasaki, T.; Wada, K.; Parhar, I.; Kamegai, J.; Sugihara, H.; Oikawa, S.; Wakabayashi, I. Generation of polyclonal antiserum against the growth hormone secretagogue receptor (GHS-R): Evidence that the GHS-R exists in the hypothalamus, pituitary and stomach of rats. Life Sci. 2001, 68, 991–996. [Google Scholar] [CrossRef]
- Zigman, J.M.; Jones, J.E.; Lee, C.E.; Saper, C.B.; Elmquist, J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 2006, 494, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Ariyasu, H.; Takaya, K.; Tagami, T.; Ogawa, Y.; Hosoda, K.; Akamizu, T.; Suda, M.; Koh, T.; Natsui, K.; Toyooka, S.; et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J. Clin. Endocrinol. Metab. 2001, 86, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Tschop, M.; Robinson, S.M.; Heiman, M.L. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 2002, 302, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, M.; Langlet, F.; Lafont, C.; Molino, F.; Hodson, D.J.; Roux, T.; Lamarque, L.; Verdié, P.; Bourrier, E.; Dehouck, B.; et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef]
- Furness, J.B.; Hunne, B.; Matsuda, N.; Yin, L.; Russo, D.; Kato, I.; Fujimiya, M.; Patterson, M.; McLeod, J.; Andrews, Z.B.; et al. Investigation of the presence of ghrelin in the central nervous system of the rat and mouse. Neuroscience 2011, 193, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.S.; Date, Y.; Yamaguchi, H.; Toshinai, K.; Tsuruta, T.; Kangawa, K.; Nakazato, M. Identification of ghrelin and its receptor in neurons of the rat arcuate nucleus. Regul. Pept. 2005, 126, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Sakata, I.; Nakano, Y.; Osborne-Lawrence, S.; Rovinsky, S.A.; Lee, C.E.; Perello, M.; Anderson, J.G.; Coppari, R.; Xiao, G.; Lowell, B.B.; et al. Characterization of a novel ghrelin cell reporter mouse. Regul. Pept. 2009, 155, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Purnell, J.Q.; Frayo, R.S.; Schmidova, K.; Wisse, B.E.; Weigle, D.S. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 2001, 50, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol. Behav. 2006, 89, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000, 141, 4255–4261. [Google Scholar] [PubMed]
- Dornonville de la Cour, C.; Bjorkqvist, M.; Sandvik, A.K.; Bakke, I.; Zhao, C.M.; Chen, D.; Hakanson, R. A-like cells in the rat stomach contain ghrelin and do not operate under gastrin control. Regul. Pept. 2001, 99, 141–150. [Google Scholar] [CrossRef]
- Drazen, D.L.; Vahl, T.P.; D’Alessio, D.A.; Seeley, R.J.; Woods, S.C. Effects of a fixed meal pattern on ghrelin secretion: Evidence for a learned response independent of nutrient status. Endocrinology 2006, 147, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Gualillo, O.; Caminos, J.E.; Kojima, M.; Kangawa, K.; Arvat, E.; Ghigo, E.; Casanueva, F.F.; Diéguez, C. Gender and gonadal influences on ghrelin mRNA levels in rat stomach. Eur. J. Endocrinol. 2001, 144, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, P.; Hassouna, R.; Grouselle, D.; Epelbaum, J.; Tolle, V. Physiological roles of preproghrelin-derived peptides in gh secretion and feeding. Peptides 2011, 32, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Verbaeys, I.; Tolle, V.; Swennen, Q.; Zizzari, P.; Buyse, J.; Epelbaum, J.; Cokelaere, M. Scheduled feeding results in adipogenesis and increased acylated ghrelin. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E1103–E1111. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, H.; Takenoya, F.; Shiba, K.; Shioda, S. Neuronal circuits involving ghrelin in the hypothalamus-mediated regulation of feeding. Neuropeptides 2010, 44, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Vodnik, M.; Strukelj, B.; Lunder, M. Ghrelin receptor ligands reaching clinical trials: From peptides to peptidomimetics; from agonists to antagonists. Horm. Metab. Res. 2016, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Inui, A.; Kaga, T.; Katsuura, G.; Fujimiya, M.; Fujino, M.A.; Kasuga, M. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut 2003, 52, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Perello, M.; Sakata, I.; Birnbaum, S.; Chuang, J.C.; Osborne-Lawrence, S.; Rovinsky, S.A.; Woloszyn, J.; Yanagisawa, M.; Lutter, M.; Zigman, J.M. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol. Psychiatry 2010, 67, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Zigman, J.M.; Nakano, Y.; Coppari, R.; Balthasar, N.; Marcus, J.N.; Lee, C.E.; Jones, J.E.; Deysher, A.E.; Waxman, A.R.; White, R.D.; et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J. Clin. Investig. 2005, 115, 3564–3572. [Google Scholar] [CrossRef] [PubMed]
- Gualillo, O.; Lago, F.; Casanueva, F.F.; Dieguez, C. One ancestor, several peptides. post-translational modifications of preproghrelin generate several peptides with antithetical effects. Mol. Cell. Endocrinol. 2006, 256, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lagaud, G.J.; Young, A.; Acena, A.; Morton, M.F.; Barrett, T.D.; Shankley, N.P. Obestatin reduces food intake and suppresses body weight gain in rodents. Biochem. Biophys. Res. Commun. 2007, 357, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, G.; Cabral, A.; Cornejo, M.P.; de Francesco, P.N.; Garcia-Romero, G.; Reynaldo, M.; Perello, M. Des-acyl ghrelin directly targets the arcuate nucleus in a ghrelin-receptor independent manner and impairs the orexigenic effect of ghrelin. J. Neuroendocrinol. 2016, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Inhoff, T.; Mönnikes, H.; Noetzel, S.; Stengel, A.; Goebel, M.; Dinh, Q.T.; Riedl, A.; Bannert, N.; Wisser, A.S.; Wiedenmann, B.; et al. Desacyl ghrelin inhibits the orexigenic effect of peripherally injected ghrelin in rats. Peptides 2008, 29, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.V.; Ren, P.G.; Avsian-Kretchmer, O.; Luo, C.W.; Raunch, R.; Klein, C.; Hsueh, A.J. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 2005, 310, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, P.; Longchamps, R.; Epelbaum, J.; Bluet-Pajot, M.-T. Obestatin partially affects ghrelin stimulation of food intake and growth hormone secretion in rodents. Endocrinology 2007, 148, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Egerod, K.L.; Schild, E.; Vickers, S.P.; Cheetham, S.; Gerlach, L.O.; Storjohann, L.; Stidsen, C.E.; Jones, R.; Beck-Sickinger, A.G.; et al. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 2007, 148, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Annemie, V.D.; van Dam, D.; Debby, V.D.; Vergote, V.; Valentijn, V.; de Spiegeleer, B.; Bart, D.S.; Luyten, W.; Walter, L.; Schoofs, L.; et al. Central administration of obestatin fails to show inhibitory effects on food and water intake in mice. Regul. Pept. 2009, 156, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Lauwers, E.; Landuyt, B.; Arckens, L.; Schoofs, L.; Luyten, W. Obestatin does not activate orphan G protein-coupled receptor GPR39. Biochem. Biophys. Res. Commun. 2006, 351, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Chartrel, N.; Alvear-Perez, R.; Leprince, J.; Iturrioz, X.; Reaux-Le Goazigo, A.; Audiont, V.; Chomarat, P.; Coge, F.; Nosjean, O.; Rodriguez, M.; et al. Comment on “Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake”. Science 2007, 315, 766. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, M.A.; Feighner, S.D.; Pong, S.; Mckee, K.K.; Hreniuk, D.L.; Silva, M.V.; Warren, V.A.; Howard, A.D.; Ploeg, L.H.Y.; van der Heck, J.V. Ghrelin: Minimal sequence of ghrelin necessary for activation of growth. J. Med. Chem. 2000, 43, 4370–4376. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.A.; Solenberg, P.J.; Perkins, D.R.; Willency, J.A.; Knierman, M.D.; Jin, Z.; Witcher, D.R.; Luo, S.; Onyia, J.E.; Hale, J.E. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc. Natl. Acad. Sci. USA 2008, 105, 6320–6325. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 2008, 132, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Staes, E.; Absil, P.A.; Lins, L.; Brasseur, R.; Deleu, M.; Lecouturier, N.; Fievez, V.; des Rieux, A.; Mingeot-Leclercq, M.P.; Raussens, V.; et al. Acylated and unacylated ghrelin binding to membranes and to ghrelin receptor: Towards a better understanding of the underlying mechanisms. Biochim. Biophys. Acta 2010, 1798, 2102–2113. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Kola, B.; Grossman, A.; Korbonits, M. The expression of ghrelin O-acyltransferase (GOAT) in human tissues. Endocr. J. 2011, 58, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Gahete, M.D.; Córdoba-Chacón, J.; Salvatori, R.; Castaño, J.P.; Kineman, R.D.; Luque, R.M. Metabolic regulation of ghrelin O-acyltransferase (GOAT) expression in the mouse hypothalamus, pituitary, and stomach. Mol. Cell. Endocrinol. 2010, 317, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, H.; Gutierrez, J.A.; Solenberg, P.J.; Pfluger, P.T.; Czyzyk, T.A.; Willency, J.A.; Schürmann, A.; Joost, H.-G.; Jandacek, R.J.; Hale, J.E.; et al. GOAT links dietary lipids with the endocrine control of energy balance. Nat. Med. 2009, 15, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Wellman, M.; Abizaid, A. Knockdown of central ghrelin O-acyltransferase by vivo-morpholino reduces body mass of rats fed a high-fat diet. Peptides 2015, 70, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.-J.; Liang, G.; Li, R.L.; Xie, X.; Sleeman, M.W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Goldstein, J.L.; Brown, M.S. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7467–7472. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Murphy, K.G.; Le Roux, C.W.; Ghatei, M.A.; Bloom, S.R. Characterization of ghrelin-like immunoreactivity in human plasma. J. Clin. Endocrinol. Metab. 2005, 90, 2205–2211. [Google Scholar] [CrossRef] [PubMed]
- McGovern-Gooch, K.R.; Rodrigues, T.; Darling, J.E.; Sieburg, M.A.; Abizaid, A.; Hougland, J.L. Ghrelin octanoylation is completely stabilized in biological samples by alkyl fluorophosphonates. Endocrinology 2016, 157, 4330–4338. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, H.; Kojima, M.; Matsuo, H.; Kangawa, K. Ghrelin and des-acyl ghrelin: Two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem. Biophys. Res. Commun. 2000, 279, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Dave, N.; Mugundu, G.M.; Davis, H.W.; Gaylinn, B.D.; Thorner, M.O.; Tschöp, M.H.; D’Alessio, D.; Desai, P.B. The pharmacokinetics of acyl, des-acyl, and total ghrelin in healthy human subjects. Eur. J. Endocrinol. 2013, 168, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Gauna, C.; van de Zande, B.; van Kerkwijk, A.; Themmen, A.P.N.; van der Lely, A.J.; Delhanty, P.J.D. Unacylated ghrelin is not a functional antagonist but a full agonist of the type 1a growth hormone secretagogue receptor (GHS-R). Mol. Cell. Endocrinol. 2007, 274, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Abizaid, A. Driving the need to feed: Insight into the collaborative interaction between ghrelin and endocannabinoid systems in modulating brain reward systems. Neurosci. Biobehav. Rev. 2016, 66, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Abizaid, A.; Horvath, T.L. Brain circuits regulating energy homeostasis. Regul. Pept. 2008, 149, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Willesen, M.G.; Kristensen, P.; Rømer, J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 1999, 70, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.W.; Elmquist, J.K. From neuroanatomy to behavior: Central integration of peripheral signals regulating feeding behavior. Nat. Neurosci. 2012, 15, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Currie, P.J.; Khelemsky, R.; Rigsbee, E.M.; Dono, L.M.; Coiro, C.D.; Chapman, C.D.; Hinchcliff, K. Ghrelin is an orexigenic peptide and elicits anxiety-like behaviors following administration into discrete regions of the hypothalamus. Behav. Brain Res. 2012, 226, 96–105. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Lage, R.; Saha, A.K.; Pérez-Tilve, D.; Vázquez, M.J.; Varela, L.; Sangiao-Alvarellos, S.; Tovar, S.; Raghay, K.; Rodríguez-Cuenca, S.; et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 2008, 7, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, P.K.; Li, D.; Grace, M.K.; Billington, C.J.; Kotz, C.M.; Levine, A.S. Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides 2003, 24, 597–602. [Google Scholar] [CrossRef]
- Abizaid, A.; Gao, Q.; Horvath, T.L. Thoughts for food: Brain mechanisms and peripheral energy balance. Neuron 2006, 51, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Cheunsuang, O.; Morris, R. Astrocytes in the arcuate nucleus and median eminence that take up a fluorescent dye from the circulation express leptin receptors and neuropeptide Y Y1 receptors. Glia 2005, 52, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Münzberg, H. Differential leptin access into the brain—A hierarchical organization of hypothalamic leptin target sites? Physiol. Behav. 2008, 94, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Norsted, E.; Gomuc, B.; Meister, B. Protein components of the blood-brain barrier (BBB) in the mediobasal hypothalamus. J. Chem. Neuroanat. 2008, 36, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Ferguson, A.V. Ghrelin: Central nervous system sites of action in regulation of energy balance. Int. J. Pept. 2010. [Google Scholar] [CrossRef] [PubMed]
- Scott, V.; McDade, D.M.; Luckman, S.M. Rapid changes in the sensitivity of arcuate nucleus neurons to central ghrelin in relation to feeding status. Physiol. Behav. 2007, 90, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.M.; Rosenzweig, I.; Dickson, S.L. Activation of arcuate nucleus neurons by systemic administration of leptin and growth hormone-releasing peptide-6 in normal and fasted rats. Neuroendocrinology 1999, 70, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hewson, A.K.; Dickson, S.L. Systemic administration of ghrelin induces Fos and Egr-1 proteins in the hypothalamic arcuate nucleus of fasted and fed rats. J. Neuroendocrinol. 2000, 12, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Bugarith, K.; Dinh, T.T.; Li, A.J.; Speth, R.C.; Ritter, S. Basomedial hypothalamic injections of neuropeptide y conjugated to saporin selectively disrupt hypothalamic controls of food intake. Endocrinology 2005, 146, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Kamegai, J.; Shimizu, T.; Ishii, S.; Sugihara, H.; Oikawa, S. Ghrelin stimulates GH but not food intake in arcuate nucleus ablated rats. Endocrinology 2002, 143, 3268–3275. [Google Scholar] [CrossRef] [PubMed]
- Kamegai, J.; Tamura, H.; Shimizu, T.; Ishii, S.; Sugihara, H.; Wakabayahi, I. Chronic central infusion of ghrelin increases hypothalamic neuropeptide y and agouti-related protein mRNA levels and body weight in rats. Diabetes 2001, 50, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Aponte, Y.; Atasoy, D.; Sternson, S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 2011, 14, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Boston, B.A.; Blaydon, K.M.; Varnerin, J.; Cone, R.D. Independent and additive effects of central POMC and leptin pathways on murine obesity. Science 1997, 278, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Ellacott, K.L.J.; Cone, R.D. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Prog. Horm. Res. 2004, 59, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.M.; Breininger, J.F.; Baskin, D.G.; Schwartz, M.W. Coexpression of AGRP and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1998, 1, 271–272. [Google Scholar] [PubMed]
- Dietrich, M.O.; Horvath, T.L. Hypothalamic control of energy balance: Insights into the role of synaptic plasticity. Trends Neurosci. 2013, 36, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L. Synaptic plasticity in energy balance regulation. Obesity 2006, 14, 228S–233S. [Google Scholar] [CrossRef] [PubMed]
- Riediger, T.; Traebert, M.; Schmid, H.A.; Scheel, C.; Lutz, T.A.; Scharrer, E. Site-specific effects of ghrelin on the neuronal activity in the hypothalamic arcuate nucleus. Neurosci. Lett. 2003, 341, 151–155. [Google Scholar] [CrossRef]
- Mountjoy, K.G.; Mortrud, M.T.; Low, M.J.; Simerly, R.B.; Cone, R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endocrinol. 1994, 8, 1298–1308. [Google Scholar] [PubMed]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Stanley, B.G.; Leibowitz, S.F. Neuroreptide Y: Stimulation of feeding and drinking by injection into the paraventricular nucleus. Life Sci. 1984, 35, 2635–2642. [Google Scholar] [CrossRef]
- Tong, Q.; Ye, C.-P.; Jones, J.E.; Elmquist, J.K.; Lowell, B.B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 2008, 11, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Adan, R.A.; Cone, R.D.; Burbach, J.P.; Gispen, W.H. Differential effects of melanocortin peptides on neural melanocortin receptors. Mol. Pharmacol. 1994, 46, 1182–1190. [Google Scholar] [PubMed]
- Biebermann, H.; Castaneda, T.R.; van Landeghem, F.; von Deimling, A.; Escher, F.; Brabant, G.; Hebebrand, J.; Hinney, A.; Tschop, M.H.; Gruters, A.; et al. A role for β-melanocyte-stimulating hormone in human body-weight regulation. Cell Metab. 2006, 3, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Elias, C.F.; Aschkenasi, C.; Lee, C.; Kelly, J.; Ahima, R.S.; Bjorbæk, C.; Flier, J.S.; Saper, C.B.; Elmquist, J.K. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 1999, 23, 775–786. [Google Scholar] [CrossRef]
- Sahm, U.G.; Qarawi, M.A.; Olivier, G.W.J.; Ahmed, A.R.H.; Branch, S.K.; Moss, S.H.; Pouton, C.W. The melanocortin (MC3) receptor from rat hypothalamus: Photoaffinity labelling and binding of alanine-substituted α-MSH analogues. FEBS Lett. 1994, 350, 29–32. [Google Scholar] [CrossRef]
- Baskin, D.G.; Breininger, J.F.; Schwartz, M.W. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes 1999, 48, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Horvath, T.L. Neurobiology of feeding and energy expenditure. Annu. Rev. Neurosci. 2007, 30, 367–398. [Google Scholar] [CrossRef] [PubMed]
- Abizaid, A. Ghrelin and dopamine: New insights on the peripheral regulation of appetite. J. Neuroendocrinol. 2009, 21, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Perelló, M.; Zigman, J.M. The role of ghrelin in reward-based eating. Biol. Psychiatry 2012, 72, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Skibicka, K.P.; Hansson, C.; Alvarez-Crespo, M.; Friberg, P.A.; Dickson, S.L. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience 2011, 180, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Disse, E.; Bussier, A.L.; Veyrat-Durebex, C.; Deblon, N.; Pfluger, P.T.; Tschöp, M.H.; Laville, M.; Rohner-Jeanrenaud, F. Peripheral ghrelin enhances sweet taste food consumption and preference, regardless of its caloric content. Physiol. Behav. 2010, 101, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Egecioglu, E.; Jerlhag, E.; Salomé, N.; Skibicka, K.P.; Haage, D.; Bohlooly-Y, M.; Andersson, D.; Bjursell, M.; Perrissoud, D.; Engel, J.A.; et al. Ghrelin increases intake of rewarding food in rodents. Addict. Biol. 2010, 15, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Landgren, S.; Simms, J.A.; Thelle, D.S.; Strandhagen, E.; Bartlett, S.E.; Engel, J.A.; Jerlhag, E. The ghrelin signalling system is involved in the consumption of sweets. PLoS ONE 2011, 6, e18170. [Google Scholar] [CrossRef] [PubMed]
- Shimbara, T.; Mondal, M.S.; Kawagoe, T.; Toshinai, K.; Koda, S.; Yamaguchi, H.; Date, Y.; Nakazato, M. Central administration of ghrelin preferentially enhances fat ingestion. Neurosci. Lett. 2004, 369, 75–79. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Rodrigues, T.; Watts, A.; Murray, E.; Wilson, A.; Abizaid, A. Investigation of a role for ghrelin signaling in binge-like feeding in mice under limited access to high-fat diet. Neuroscience 2016, 319, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Finger, B.C.; Dinan, T.G.; Cryan, J.F. Diet-induced obesity blunts the behavioural effects of ghrelin: Studies in a mouse-progressive ratio task. Psychopharmacology 2012, 220, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Baler, R.D.; Volkow, N.D. Drug addiction: The neurobiology of disrupted self-control. Trends Mol. Med. 2006, 12, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B.; Chou, T.C.; Elmquist, J.K. The need to feed: Homeostatic and hedonic control of eating. Neuron 2002, 36, 199–211. [Google Scholar] [CrossRef]
- Salamone, J.D.; Zigmond, M.J.; Stricker, E.M. Characterization of the impaired feeding behavior in rats given haloperidol or dopamine-depleting brain lesions. Neuroscience 1990, 39, 17–24. [Google Scholar] [CrossRef]
- Salamone, J.D.; Mahan, K.; Rogers, S. Ventrolateral striatal dopamine depletions impair feeding and food handling in rats. Pharmacol. Biochem. Behav. 1993, 44, 605–610. [Google Scholar] [CrossRef]
- Richardson, N.R.; Gratton, A. Behavior-relevant changes in nucleus accumbens dopamine transmission elicited by food reinforcement: An electrochemical study in rat. J. Neurosci. 1996, 16, 8160–8169. [Google Scholar] [PubMed]
- Naleid, A.M.; Grace, M.K.; Cummings, D.E.; Levine, A.S. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides 2005, 26, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Isaacs, A.M.; O’Farrell, E.; Abizaid, A. Motivation to obtain preferred foods is enhanced by ghrelin in the ventral tegmental area. Horm. Behav. 2011, 60, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, Z.Y.; Nicholson, M.L.; Currie, P.J. 6-hydroxydopamine lesions of the ventral tegmental area suppress ghrelin’s ability to elicit food-reinforced behavior. Neurosci. Lett. 2011, 499, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Skibicka, K.P.; Shirazi, R.H.; Rabasa-Papio, C.; Alvarez-Crespo, M.; Neuber, C.; Vogel, H.; Dickson, S.L. Divergent circuitry underlying food reward and intake effects of ghrelin: Dopaminergic VTA-accumbens projection mediates ghrelin’s effect on food reward but not food intake. Neuropharmacology 2013, 73, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Cone, J.J.; Roitman, J.D.; Roitman, M.F. Ghrelin regulates phasic dopamine and nucleus accumbens signaling evoked by food-predictive stimuli. J. Neurochem. 2015, 133, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Jerlhag, E.; Egecioglu, E.; Dickson, S.L.; Douhan, A.; Svensson, L.; Engel, J.A. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict. Biol. 2007, 12, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Skibicka, K.P.; Hansson, C.; Egecioglu, E.; Dickson, S.L. Role of ghrelin in food reward: Impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict. Biol. 2012, 17, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Kaneko, F.; Yamada, M.; Kishikawa, Y.; Kawahara, H.; Nishi, A. Food reward-sensitive interaction of ghrelin and opioid receptor pathways in mesolimbic dopamine system. Neuropharmacology 2013, 67, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Perello, M.; Sakata, I.; Osborne-Lawrence, S.; Savitt, J.M.; Lutter, M.; Zigman, J.M. Ghrelin mediates stress-induced food-reward behavior in mice. J. Clin. Investig. 2011, 121, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Cabral, A.; Valdivia, S.; Fernandez, G.; Reynaldo, M.; Perello, M. Divergent neuronal circuitries underlying acute orexigenic effects of peripheral or central ghrelin: Critical role of brain accessibility. J. Neuroendocrinol. 2014, 26, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Murakami, N.; Toshinai, K.; Matsukura, S.; Niijima, A.; Matsuo, H.; Kangawa, K.; Nakazato, M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 2002, 123, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.M.; Sharp, F.R.; Curran, T. Expression of c-Fos protein in brain: Metabolic mapping at the cellular level. Science 1988, 240, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Rüter, J.; Kobelt, P.; Tebbe, J.J.; Avsar, Y.; Veh, R.; Wang, L.; Klapp, B.F.; Wiedenmann, B.; Taché, Y.; Mönnikes, H. Intraperitoneal injection of ghrelin induces Fos expression in the paraventricular nucleus of the hypothalamus in rats. Brain Res. 2003, 991, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Traebert, M.; Riediger, T.; Whitebread, S.; Scharrer, E.; Schmid, H.A. Ghrelin acts on leptin-responsive neurones in the rat arcuate nucleus. J. Neuroendocrinol. 2002, 14, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Saint-Pierre, D.H.; Taché, Y. Peripheral ghrelin selectively increases Fos expression in neuropeptide Y—Synthesizing neurons in mouse hypothalamic arcuate nucleus. Neurosci. Lett. 2002, 325, 47–51. [Google Scholar] [CrossRef]
- Hashimoto, H.; Fujihara, H.; Kawasaki, M.; Saito, T.; Shibata, M.; Otsubo, H.; Takei, Y.; Ueta, Y. Centrally and peripherally administered ghrelin potently inhibits water intake in rats. Endocrinology 2007, 148, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, X.; Zhao, Y.; Chen, S.; Owyang, C. Ghrelin acts on the dorsal vagal complex to stimulate pancreatic protein secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1350–G1358. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Johno, Y.; Hayashi, K.; Yakabi, K.; Tanaka, T.; Ro, S. Expression of c-Fos protein in the brain after intravenous injection of ghrelin in rats. Neurosci. Lett. 2007, 417, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, P.; Wisser, A.S.; Stengel, A.; Goebel, M.; Inhoff, T.; Noetzel, S.; Veh, R.W.; Bannert, N.; van der Voort, I.; Wiedenmann, B.; et al. Peripheral injection of ghrelin induces Fos expression in the dorsomedial hypothalamic nucleus in rats. Brain Res. 2008, 1204, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lin, L.; Xu, P.; Saito, K.; Wei, Q.; Meadows, A.G.; Bongmba, O.Y.N.; Pradhan, G.; Zheng, H.; Xu, Y.; et al. Neuronal deletion of ghrelin receptor almost completely prevents diet-induced obesity. Diabetes 2016, 65, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- McGirr, R.; McFarland, M.S.; McTavish, J.; Luyt, L.G.; Dhanvantari, S. Design and characterization of a fluorescent ghrelin analog for imaging the growth hormone secretagogue receptor 1a. Regul. Pept. 2011, 172, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.A.; Bauer, B.; Fahlke, C.; Fricker, G.; Iadecola, C.; Janigro, D.; Leybaert, L.; Molnár, Z.; O’Donnell, M.E.; Povlishock, J.T.; et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat. Rev. Neurosci. 2011, 12, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell. Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vasc. Pharmacol. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Brain meets body: The blood-brain barrier as an endocrine interface. Endocrinology 2012, 153, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- M, C.; Braun, L.D.; Oldendorf, W.H.; Hill, M.A. Comparison of lipid mediated blood brain barrier penetrability in neo nates and adults. Am. J. Physiol. 1982, 243, C161–C168. [Google Scholar]
- Banks, W.A.; Kastin, A.J. Passage of peptides across the blood-brain barrier: Pathophysiological perspectives. Life Sci. 1996, 59, 1923–1943. [Google Scholar] [CrossRef]
- Pan, W.; Tu, H.; Kastin, A.J. Differential BBB interactions of three ingestive peptides: Obestatin, ghrelin, and adiponectin. Peptides 2006, 27, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Harrold, J.A.; Dovey, T.; Cai, X.-J.; Halford, J.C.G.; Pinkney, J. Autoradiographic analysis of ghrelin receptors in the rat hypothalamus. Brain Res. 2008, 1196, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Goebel, M.; Wang, L.; Taché, Y.; Sachs, G.; Lambrecht, N.W.G. Differential distribution of ghrelin-O-acyltransferase (GOAT) immunoreactive cells in the mouse and rat gastric oxyntic mucosa. Biochem. Biophys. Res. Commun. 2010, 392, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.P.; Prechtl, C.G.; Scholtz, S.; Miras, A.D.; Chhina, N.; Durighel, G.; Deliran, S.S.; Beckmann, C.; Ghatei, M.A.; Ashby, D.R.; et al. Ghrelin mimics fasting to enhance human hedonic, orbitofrontal cortex, and hippocampal responses to food. Am. J. Clin. Nutr. 2014, 99, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, N.B.; Krebs, L.; Kobiella, A.; Grimm, O.; Pilhatsch, M.; Bidlingmaier, M.; Zimmermann, U.S.; Smolka, M.N. Fasting levels of ghrelin covary with the brain response to food pictures. Addict. Biol. 2013, 18, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; McGlone, F.; Bedrossian, D.; Dagher, A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008, 7, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Chollet, C.; Bergmann, R.; Pietzsch, J.; Beck-Sickinger, A.G. Design, evaluation, and comparison of ghrelin receptor agonists and inverse agonists as suitable radiotracers for PET imaging. Bioconjug. Chem. 2012, 23, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Sawchenko, P.E. Central connections of the sensory and motor nuclei of the vagus nerve. J. Auton. Nerv. Syst. 1983, 9, 13–26. [Google Scholar] [CrossRef]
- Van der Kooy, D.; Koda, L.Y.; McGinty, J.F.; Gerfen, C.R.; Bloom, F.E. The organization of projections from the cortex, amygdala, and hypothalamus to the nucleus of the solitary tract in rat. J. Comp. Neurol. 1984, 224, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Agostoni, E.; Chinnock, J.E.; Daly, M.D.B.; Murray, J.G. Functional and histological studies of the vagus nerve and its branches to the heart, lungs and abdominal viscera in the cat. J. Physiol. 1957, 135, 182–205. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Shimbara, T.; Koda, S.; Toshinai, K.; Ida, T.; Murakami, N.; Miyazato, M.; Kokame, K.; Ishizuka, Y.; Ishida, Y.; et al. Peripheral ghrelin transmits orexigenic signals through the noradrenergic pathway from the hindbrain to the hypothalamus. Cell Metab. 2006, 4, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Woods, S.C.; Porte, D.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [PubMed]
- Asakawa, A.; Inui, A.; Kaga, T.; Yuzuriha, H.; Nagata, T.; Ueno, N.; Makino, S.; Fujimiya, M.; Niijima, A.; Fujino, M.A.; et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology 2001, 120, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, C.W.; Neary, N.M.; Halsey, T.J.; Small, C.J.; Martinez-Isla, A.M.; Ghatei, M.A.; Theodorou, N.A.; Bloom, S.R. Ghrelin does not stimulate food intake in patients with surgical procedures involving vagotomy. J. Clin. Endocrinol. Metab. 2005, 90, 4521–4524. [Google Scholar] [CrossRef] [PubMed]
- Sakata, I.; Yamazaki, M.; Inoue, K.; Hayashi, Y.; Kangawa, K.; Sakai, T. Growth hormone secretagogue receptor expression in the cells of the stomach-projected afferent nerve in the rat nodose ganglion. Neurosci. Lett. 2003, 342, 183–186. [Google Scholar] [CrossRef]
- Page, A.J.; Slattery, J.A.; Milte, C.; Laker, R.; O’Donnell, T.; Dorian, C.; Brierley, S.M.; Blackshaw, L.A. Ghrelin selectively reduces mechanosensitivity of upper gastrointestinal vagal afferents. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1376–G1384. [Google Scholar] [CrossRef] [PubMed]
- Sawchenko, P.E.; Swanson, L.W. Central noradrenergic pathways for the integration of hypothalamic neuroendocrine and autonomic responses. Science 1981, 214, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Mura, A.; Langhans, W.; Geary, N. Gut vagal afferents are not necessary for the eating-stimulatory effect of intraperitoneally injected ghrelin in the rat. J. Neurosci. 2006, 26, 11052–11060. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.N.; Kiss, J.Z.; Mezey, E. Bilateral midbrain transections block the behavioral effects of cholecystokinin on feeding and exploration in rats. Brain Res. 1984, 322, 316–321. [Google Scholar] [CrossRef]
- Kentish, S.; Li, H.; Philp, L.K.; O’Donnell, T.A.; Isaacs, N.J.; Young, R.L.; Wittert, G.A.; Blackshaw, L.A.; Page, A.J. Diet-induced adaptation of vagal afferent function. J. Physiol. 2012, 590, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Kentish, S.J.; O’Donnell, T.A.; Isaacs, N.J.; Young, R.L.; Li, H.; Harrington, A.M.; Brierley, S.M.; Wittert, G.A.; Blackshaw, L.A.; Page, A.J. Gastric vagal afferent modulation by leptin is influenced by food intake status. J. Physiol. 2013, 591, 1921–1934. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.M. Morphology and physiology of capillary systems in subregions of the subfornical organ and area postrema. Can. J. Physiol. Pharmacol. 1991, 69, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.M.; Weindl, A. Peering through the windows of the brain. J. Cereb. Blood Flow Metab. 1987, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- McKinley, M.J.; McAllen, R.M.; Davern, P.; Giles, M.E.; Penschow, J.; Sunn, N.; Uschakov, A.; Oldfield, B.J. The sensory circumventricular organs of the mammalian brain. Adv. Anat. Embryol. Cell Biol. 2003, 172, 1–122. [Google Scholar]
- Banks, W.A. The blood-brain barrier: Connecting the gut and the brain. Regul. Pept. 2008, 149, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Broadwell, R.D.; Brightman, M.W. Entry of peroxidase into neurons of the central and peripheral nervous systems from extracerebral and cerebral blood. J. Comp. Neurol. 1976, 166, 257–283. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Dreifuss, J.-J.; Dziegielewska, K.M.; Johansson, P.A.; Habgood, M.D.; Møllgård, K.; Bauer, H.-C. The rights and wrongs of blood-brain barrier permeability studies: A walk through 100 years of history. Front. Neurosci. 2014, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Bolborea, M.; Dale, N. Hypothalamic tanycytes: Potential roles in the control of feeding and energy balance. Trends Neurosci. 2013, 36, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Langlet, F.; Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Tanycyte-like cells form a blood-cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. J. Comp. Neurol. 2013, 521, 3389–3405. [Google Scholar] [CrossRef] [PubMed]
- Langlet, F.; Levin, B.E.; Luquet, S.; Mazzone, M.; Messina, A.; Dunn-meynell, A.A.; Balland, E.; Lacombe, A.; Mazur, D.; Carmeliet, P.; et al. Tanycytic VEGF-A Boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab. 2013, 17, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Duvernoy, H.M.; Risold, P.Y. The circumventricular organs: An atlas of comparative anatomy and vascularization. Brain Res. Rev. 2007, 56, 119–147. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Ferguson, A.V. The sensory circumventricular organs: Brain targets for circulating signals controlling ingestive behavior. Physiol. Behav. 2007, 91, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Shaver, S.; Pang, J.J.; Wainman, D.S.; Wall, K.M.; Gross, P.M. Morphology and function of capillary networks in subregions of the rat tuber cinereum. Cell Tissue Res. 1992, 267, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Ambach, G.; Palkovits, M.; Szentagothai, J. blood supply of the rat hypothalamus. IV. Retrochiasmatic area, median eminence, arcuate nucleus. Acta Morphol. Acad. Sci. Hung. 1976, 24, 93–119. [Google Scholar] [PubMed]
- Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J. Comp. Neurol. 2010, 518, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, S.R.; Lomniczi, A.; Sandau, U.S. Glial-gonadotrophin hormone (GnRH) neurone interactions in the median eminence and the control of GnRH Secretion. J. Neuroendocrinol. 2008, 20, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.M.; Blázquez, J.L.; Pastor, F.E.; Peláez, B.; Peña, P.; Peruzzo, B.; Amat, P. Hypothalamic tanycytes: A key component of brain-endocrine interaction. Int. Rev. Cytol. 2005, 247, 89–164. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Sanders, N.M.; Compton, D.; Becker, T.C.; Eiki, J.I.; Zhang, B.B.; Levin, B.E. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J. Neurosci. 2009, 29, 7015–7022. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, B.; Pastor, F.E.; Blázquez, J.L.; Amat, P.; Rodríguez, E.M. Polarized endocytosis and transcytosis in the hypothalamic tanycytes of the rat. Cell Tissue Res. 2004, 317, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Cheunsuang, O.; Stewart, A.L.; Morris, R. Differential uptake of molecules from the circulation and CSF reveals regional and cellular specialisation in CNS detection of homeostatic signals. Cell Tissue Res. 2006, 325, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, P.; Leroy, D.; Tramu, G. Sexual dimorphism in the organization of the rat hypothalamic infundibular area. Neuroscience 2006, 141, 1731–1745. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, P.; Garret, M.; Lapirot, O.; Lafon, P.; Loyens, A.; Prevot, V.; Levine, J.E. Brain-endocrine interactions: A microvascular route in the mediobasal hypothalamus. Endocrinology 2009, 150, 5509–5519. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.P.; Dube, M.G.; Pu, S.; Xu, B.; Horvath, T.L.; Kalra, P.S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 1999, 20, 68–100. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The Many Lives of Leptin. Peptides 2004, 25, 331–338. [Google Scholar] [CrossRef] [PubMed]
- De Lange, E.C.M.; Bouw, M.R.; Mandema, J.W.; Danhof, M.; de Boer, A.G.; Breimer, D.D. Application of intracerebral microdialysis to study regional distribution kinetics of drugs in rat brain. Br. J. Pharmacol. 1995, 116, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, B.; Pastor, F.E.; Blazquez, J.L.; Schobitz, K.; Pelaez, B.; Amat, P.; Rodríguez, E.M. A second look at the barriers of the medial basal hypothalamus. Exp. Brain Res. 2000, 132, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Réthelyi, M. Diffusional barrier around the hypothalamic arcuate nucleus in the rat. Brain Res. 1984, 307, 355–358. [Google Scholar] [CrossRef]
- Cabral, A.; Fernandez, G.; Perello, M. Analysis of brain nuclei accessible to ghrelin present in the cerebrospinal fluid. Neuroscience 2013, 253, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Mimee, A.; Smith, P.M.; Ferguson, A.V. Circumventricular organs: Targets for integration of circulating fluid and energy balance signals? Physiol. Behav. 2013, 121, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.T.; Ferguson, A.V. Sensory circumventricular organs: Central roles in integrated autonomic regulation. Regul. Pept. 2004, 117, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.V.; Day, T.A.; Renaud, L.P. Subfornical organ efferents influence the excitability of neurohypophyseal and tuberoinfundibular paraventricular nucleus neurons in the rat. Neuroendocrinology 1984, 39, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Gruber, K.; McRae-Degueurce, A.; Wilkin, L.D.; Mitchell, L.D.; Johnson, A.K. Forebrain and brainstem afferents to the arcuate nucleus in the rat: Potential pathways for the modulation of hypophyseal secretions. Neurosci. Lett. 1987, 75, 1–5. [Google Scholar] [CrossRef]
- Lind, R.W.; van Hoesen, G.W.; Johnson, A.K. An HRP study of the connections of the subfornical organ of the rat. J. Comp. Neurol. 1982, 210, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Miselis, R.R.; Shapiro, R.E.; Hand, P.J. Subfornical organ efferents to neural systems for control of body water. Science 1979, 205, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Sgro, S.; Ferguson, A.V.; Renaud, L.P. Subfornical organ-supraoptic nucleus connections: An electrophysiologic study in the rat. Brain Res. 1984, 303, 7–13. [Google Scholar] [CrossRef]
- Hoyda, T.D.; Smith, P.M.; Ferguson, A.V. Gastrointestinal hormone actions in the central regulation of energy metabolism: Potential sensory roles for the circumventricular organs. Int. J. Obes. 2009, 33, S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Miselis, R.R. The efferent projections of the subfornical organ of the rat: A circumventricular organ within a neural network subserving water balance. Brain Res. 1981, 230, 1–23. [Google Scholar] [CrossRef]
- Pulman, K.J.; Fry, W.M.; Cottrell, G.T.; Ferguson, A.V. The Subfornical organ: A central target for circulating feeding signals. J. Neurosci. 2006, 26, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.E.; Miselis, R.R. The central neural connections of the area postrema of the rat. J. Comp. Neurol. 1985, 234, 344–364. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooy, D.; Koda, L.Y. Organization of the projections of a circumventricular organ: The area postrema in the rat. J. Comp. Neurol. 1983, 219, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Vigier, D.; Rouviere, A. Afferent and efferent connections of the area postrema demonstrated by the horseradish peroxidase method. Arch. Ital. Biol. 1979, 117, 325–339. [Google Scholar] [PubMed]
- Faulconbridge, L.F.; Cummings, D.E.; Kaplan, J.M.; Grill, H.J. Hyperphagic effects of brainstem ghrelin administration. Diabetes 2003, 52, 2260–2265. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Ferguson, A.V. Ghrelin modulates electrical activity of area postrema neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.R.T.; Von Englehardt, N.; Leng, G.; Smith, R.G.; Dickson, S.L. Growth hormone secretagogue activation of the arcuate nucleus and brainstem occurs via a non-noradrenergic pathway. J. Neuroendocrinol. 2000, 12, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Gilg, S.; Lutz, T.A. The orexigenic effect of peripheral ghrelin differs between rats of different age and with different baseline food intake, and it may in part be mediated by the area postrema. Physiol. Behav. 2006, 87, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Faulconbridge, L.F.; Grill, H.J.; Kaplan, J.M.; Daniels, D. Caudal brainstem delivery of ghrelin induces fos expression in the nucleus of the solitary tract, but not in the arcuate or paraventricular nuclei of the hypothalamus. Brain Res. 2008, 1218, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.B.; Snape, A.C.; Baudoin, F.M.H.; Luckman, S.M. Acute central ghrelin and gh secretagogues induce feeding and activate brain appetite centers. Endocrinology 2002, 143, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Grouselle, D.; Chaillou, E.; Caraty, A.; Bluet-Pajot, M.-T.; Zizzari, P.; Tillet, Y.; Epelbaum, J. Pulsatile cerebrospinal fluid and plasma ghrelin in relation to growth hormone secretion and food intake in the sheep. J. Neuroendocrinol. 2008, 20, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Tritos, N.A.; Kokkinos, A.; Lampadariou, E.; Alexiou, E.; Katsilambros, N.; Maratos-Flier, E. Cerebrospinal fluid ghrelin is negatively associated with body mass index. J. Clin. Endocrinol. Metab. 2003, 88, 2943–2946. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Cygankiewicz, A.; Jensen, T.H.; Ankersen, M.; Schwartz, T.W. high constitutive signaling of the ghrelin receptor—Identification of a potent inverse agonist. Mol. Endocrinol. 2003, 17, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nakajima, K.; Oomura, Y.; Wayner, M.J.; Sasaki, K. Electrophysiological effects of ghrelin on pedunculopontine tegmental neurons in rats: An in vitro study. Peptides 2009, 30, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Miao, Y.; Gao, L.; Pan, H.; Zhu, S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the dvc of brainstem in rat. Regul. Pept. 2006, 134, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Fukue, Y.; Teranishi, H.; Yoshida, Y.; Kojima, M. Molecular forms of hypothalamic ghrelin and its regulation by fasting and 2-deoxy-d-glucose administration. Endocrinology 2005, 146, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, I.I.; le Feber, J. Ghrelin accelerates synapse formation and activity development in cultured cortical networks. BMC Neurosci. 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Wortley, K.E.; Anderson, K.D.; Garcia, K.; Murray, J.D.; Malinova, L.; Liu, R.; Moncrieffe, M.; Thabet, K.; Cox, H.J.; Yancopoulos, G.D.; et al. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc. Natl. Acad. Sci. USA 2004, 101, 8227–8232. [Google Scholar] [CrossRef] [PubMed]
- Canpolat, S.; Aydin, M.; Yasar, A.; Colakoglu, N.; Yilmaz, B.; Kelestimur, H. Effects of pinealectomy and exogenous melatonin on immunohistochemical ghrelin staining of arcuate nucleus and serum ghrelin leves in the rat. Neurosci. Lett. 2006, 410, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Wang, Q.P.; Kageyama, H.; Takenoya, F.; Kita, T.; Matsuoka, T.; Funahashi, H.; Shioda, S. Synaptic interactions between ghrelin- and neuropeptide Y-containing neurons in the rat arcuate nucleus. Peptides 2003, 24, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Okuda, H.; Takenoya, F.; Kintaka, Y.; Yagi, M.; Wang, L.; Seki, M.; Hori, Y.; Kageyama, H.; Shioda, S. Synaptic relationships between proopiomelanocortin- and ghrelin-containing neurons in the rat arcuate nucleus. Regul. Pept. 2008, 145, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Kohno, D.; Gao, H.Z.; Muroya, S.; Kikuyama, S.; Yada, T. Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes 2003, 52, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Guan, J.L.; Wang, Q.P.; Uehara, K.; Yamada, S.; Goto, N.; Date, Y.; Nakazato, M.; Kojima, M.; Kangawa, K.; et al. Immunocytochemical observation of ghrelin-containing neurons in the rat arcuate nucleus. Neurosci. Lett. 2002, 321, 157–160. [Google Scholar] [CrossRef]
- Menyhért, J.; Wittmann, G.; Hrabovszky, E.; Szlávik, N.; Keller, É.; Tschöp, M.; Liposits, Z.; Fekete, C. Distribution of ghrelin-immunoreactive neuronal networks in the human hypothalamus. Brain Res. 2006, 1125, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, H.; Kitamura, Y.; Hosono, T.; Kintaka, Y.; Seki, M.; Takenoya, F.; Hori, Y.; Nonaka, N.; Arata, S.; Shioda, S. Visualization of ghrelin-producing neurons in the hypothalamic arcuate nucleus using ghrelin-egfp transgenic mice. Regul. Pept. 2008, 145, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.F.; Cayer, C.; Wellman, M.; Abizaid, A.; James, J.S.; Merali, Z. In Vivo Ghrelin Release from the Arcuate Nucleus Measured by Push-Pull Perfusion. Proceedings of Society for Neuroscience: 44th Annual Meeting, Washington, DC, USA, 15–19 November 2014. [Google Scholar]
- Mori, K.; Yoshimoto, A.; Takaya, K.; Hosoda, K.; Ariyasu, H.; Yahata, K.; Mukoyama, M.; Sugawara, A.; Hosoda, H.; Kojima, M.; et al. Kidney produces a novel acylated peptide, ghrelin. FEBS Lett. 2000, 486, 213–216. [Google Scholar] [CrossRef]
- Volante, M.; Allia, E.; Gugliotta, P.; Funaro, A.; Broglio, F.; Deghenghi, R.; Muccioli, G.; Ghigo, E.; Papotti, M. Expression of ghrelin and the GH secretatogue receptor by pancreatic islet cells and related endocrine tumors. J. Clin. Endocrinol. Metab. 2002, 87, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, H.; Funahashi, H.; Hirayama, M.; Takenoya, F.; Kita, T.; Kato, S.; Sakurai, J.; Lee, E.Y.; Inoue, S.; Date, Y.; et al. Morphological analysis of ghrelin and its receptor distribution in the rat pancreas. Regul. Pept. 2005, 126, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Granata, R.; Baragli, A.; Settanni, F.; Scarlatti, F.; Ghigo, E. Unraveling the role of the ghrelin gene peptides in the endocrine pancreas. J. Mol. Endocrinol. 2010, 45, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Arnes, L.; Hill, J.T.; Gross, S.; Magnuson, M.A.; Sussel, L. Ghrelin expression in the mouse pancreas defines a unique multipotent progenitor population. PLoS ONE 2012, 7, e52026. [Google Scholar] [CrossRef] [PubMed]
- De la Cour, C.D.; Norlén, P.; Håkanson, R. Secretion of ghrelin from rat stomach ghrelin cells in response to local microinfusion of candidate messenger compounds: A microdialysis study. Regul. Pept. 2007, 143, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Wellman, M.; Abizaid, A. Growth hormone secretagogue receptor dimers: A new pharmacological target(1,2,3). eNeuro 2015, 2, e0053-14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Betancourt, L.; Smith, R.G. Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol. Endocrinol. 2006, 20, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Albarran-Zeckler, R.; Walsh, H.; Smith, R. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 2012, 73, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; van Oeffelen, W.E.P.A.; Dinan, T.G.; Cryan, J.F. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. J. Biol. Chem. 2013, 288, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Dinan, T.G.; Cryan, J.F. Taking two to tango: A role for ghrelin receptor heterodimerization in stress and reward. Front. Neurosci. 2013, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, M.R.; Brown, M.S.; Goldstein, J.L.; Zhao, T.J. Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell metab. 2014, 20, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.T.; Kirchner, H.; Gunnel, S.; Schrott, B.; Perez-Tilve, D.; Fu, S.; Benoit, S.C.; Horvath, T.; Joost, H.G.; Wortley, K.E.; et al. Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G610–G618. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ahmed, S.; Smith, R.G. Deletion of ghrelin impairs neither growth nor appetite. Mol. Cell. Biol. 2003, 23, 7973–7981. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; Zigman, J.M.; Perelló, M. Ghrelin and eating behavior: Evidence and insights from genetically-modified mouse models. Front. Neurosci. 2013, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Schwartz, T.W. Constitutive ghrelin receptor activity as a signaling set-point in appetite regulation. Trends Pharmacol. Sci. 2004, 25, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.H.; Seifert, R. Coexpression systems as models for the analysis of constitutive GPCR activity. Methods Enzymol. 2010, 485, 527–557. [Google Scholar] [PubMed]
- Damian, M.; Marie, J.; Leyris, J.P.; Fehrentz, J.A.; Verdié, P.; Martinez, J.; Banères, J.L.; Mary, S. High constitutive activity is an intrinsic feature of ghrelin receptor protein: A study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J. Biol. Chem. 2012, 287, 3630–3641. [Google Scholar] [CrossRef] [PubMed]
- Els, S.; Schild, E.; Petersen, P.S.; Kilian, T.M.; Mokrosinski, J.; Frimurer, T.M.; Chollet, C.; Schwartz, T.W.; Holst, B.; Beck-Sickinger, A.G. An aromatic region to induce a switch between agonism and inverse agonism at the ghrelin receptor. J. Med. Chem. 2012, 55, 7437–7449. [Google Scholar] [CrossRef] [PubMed]
- Pantel, J.; Legendre, M.; Cabrol, S.; Hilal, L.; Hajaji, Y.; Morisset, S.; Nivot, S.; Vie-Luton, M.-P.; Grouselle, D.; de Kerdanet, M.; et al. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J. Clin. Investig. 2006, 116, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.S.; Woldbye, D.P.D.; Madsen, A.N.; Egerod, K.L.; Jin, C.; Lang, M.; Rasmussen, M.; Beck-Sickinger, A.G.; Holst, B. In vivo characterization of high basal signaling from the ghrelin receptor. Endocrinology 2009, 150, 4920–4930. [Google Scholar] [CrossRef] [PubMed]
- Lopez Soto, E.J.; Agosti, F.; Cabral, A.; Mustafa, E.R.; Damonte, V.M.; Gandini, M.A.; Rodriguez, S.; Castrogiovanni, D.; Felix, R.; Perello, M.; et al. Constitutive and ghrelin-dependent GHSR1a activation impairs CaV2.1 and CaV2.2 currents in hypothalamic neurons. J. Gen. Physiol. 2015, 146, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Yoon, C.Y.; Park, K.H.; Shin, C.S.; Park, K.S.; Kim, S.Y.; Cho, B.Y.; Lee, H.K. Changes in ghrelin and ghrelin receptor expression according to feeding status. Neuroreport 2003, 14, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Tovar, S.; Mitchell, S.E.; Rayner, D.V.; Archer, Z.A.; Dieguez, C.; Williams, L.M. Regulation of growth hormone secretagogue receptor gene expression in the arcuate nuclei of the rat by leptin and ghrelin. Diabetes 2004, 53, 2552–2558. [Google Scholar] [CrossRef] [PubMed]
- Bellot, M.; Galandrin, S.; Boularan, C.; Matthies, H.J.; Despas, F.; Denis, C.; Javitch, J.; Mazères, S.; Sanni, S.J.; Pons, V.; et al. Dual agonist occupancy of AT1-R-α2C-AR heterodimers results in atypical Gs-PKA signaling. Nat. Chem. Biol. 2015, 11, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, M. GPCRs: Heterodimer-specific signaling. Nat. Chem. Biol. 2015, 11, 244–245. [Google Scholar] [CrossRef] [PubMed]
- Prinster, S.C.; Hague, C.; Hall, R.A. Heterodimerization of G protein-coupled receptors: Specificity and functional significance. Pharmacol. Rev. 2005, 57, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Szidonya, L.; Cserzo, M.; Hunyady, L. Dimerization and oligomerization of G-protein-coupled receptors: Debated structures with established and emerging functions. J. Endocrinol. 2008, 196, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Grande, C.; Smith, R.G. Apo-ghrelin receptor (apo-GHSR1a) regulates dopamine signaling in the brain. Front. Endocrinol. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Mavrikaki, M.; Ullrich, C.; Albarran-Zeckler, R.; Faruzzi Brantley, A.; Smith, R.G. Hippocampal dopamine/DRD1 signaling dependent on the ghrelin receptor. Cell 2015, 163, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Mary, S.; Fehrentz, J.A.; Damian, M.; Gaibelet, G.; Orcel, H.; Verdié, P.; Mouillac, B.; Martinez, J.; Marie, J.; Banères, J.L. Heterodimerization with its splice variant blocks the ghrelin receptor 1a in a non-signaling conformation: A study with a purified heterodimer assembled into lipid discs. J. Biol. Chem. 2013, 288, 24656–24665. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Aguinaga, D.; Angelats, E.; Medrano, M.; Moreno, E.; Mallol, J.; Cortés, A.; Canela, E.I.; Casadó, V.; McCormick, P.J.; et al. A significant role of the truncated ghrelin receptor GHS-R1b in ghrelin-induced signaling in neurons. J. Biol. Chem. 2016, 291, 13048–13062. [Google Scholar] [CrossRef] [PubMed]
- Rediger, A.; Tarnow, P.; Bickenbach, A.; Schaefer, M.; Krude, H.; Grüters, A.; Biebermann, H. Heterodimerization of hypothalamic G-protein-coupled receptors involved in weight regulation. Obes. Facts 2009, 2, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Rediger, A.; Piechowski, C.L.; Yi, C.X.; Tarnow, P.; Strotmann, R.; Grüters, A.; Krude, H.; Schöneberg, T.; Tschöp, M.H.; Kleinau, G.; et al. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J. Biol. Chem. 2011, 286, 39623–39631. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.B.; Cheng, C.H.K. Identification and functional characterization of two alternatively spliced growth hormone secretagogue receptor transcripts from the pituitary of black seabream acanthopagrus schlegeli. Mol. Cell. Endocrinol. 2004, 214, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Dezaki, K. Ghrelin function in insulin release and glucose metabolism. Endocr. Dev. 2013, 25, 135–143. [Google Scholar] [PubMed]
- Yin, Y.; Li, Y.; Zhang, W. The growth hormone secretagogue receptor: Its intracellular signaling and regulation. Int. J. Mol. Sci. 2014, 15, 4837–4855. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Ploeg, L.H.T.; van der Howard, A.D.; Feighner, S.D.; Cheng, K.; Hickey, G.J.; Matthew, J.; Wyvratt, J.; Fisher, M.H.; Nargund, R.P.; et al. Peptidomimetic regulation of growth hormone secretion. Endocr. Rev. 2011, 18, 621–645. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Kohno, D.; Sone, H.; Yada, T. Ghrelin raises [Ca2+]i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem. Biophys. Res. Commun. 2008, 336, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Kola, B. Role of AMP-activated protein kinase in the control of appetite. J. Neuroendocrinol. 2008, 20, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.P.; Reoma, J.L.; Gamm, D.M.; Uhler, M.D. LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem. J. 2000, 345, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.A.; Langmead, C.J.; Wise, A.; Milligan, G. Growth hormone secretagogues and growth hormone releasing peptides act as orthosteric super-agonists but not allosteric regulators for activation of the G protein Gαo1 by the ghrelin receptor. Mol. Pharmacol. 2009, 76, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Dezaki, K.; Kakei, M.; Yada, T. Ghrelin uses Gαi2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet β-cells. Diabetes 2007, 56, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Clemett, D.A.; Punhani, T.; Duxon, M.S.; Blackburn, T.P.; Fone, K.C.F. Immunohistochemical localisation of the 5-HT2c receptor protein in the rat CNS. Neuropharmacology 2000, 39, 123–132. [Google Scholar] [CrossRef]
- Wright, D.E.; Seroogy, K.B.; Lundgren, K.H.; Davis, B.M.; Jennes, L. Comparative localization of serotonin, 1a, 1c and 2 receptor subtype mRNAs in rat brain. J. Comp. Neurol. 1995, 351, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Nonogaki, K.; Ohashi-Nozue, K.; Oka, Y. A negative feedback system between brain serotonin systems and plasma active ghrelin levels in mice. Biochem. Biophys. Res. Commun. 2006, 341, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Currie, P.J.; Coiro, C.D.; Niyomchai, T.; Lira, A.; Farahmand, F. Hypothalamic paraventricular 5-hydroxytryptamine: Receptor-specific inhibition of NPY-stimulated eating and energy metabolism. Pharmacol. Biochem. Behav. 2002, 71, 709–716. [Google Scholar] [CrossRef]
- Currie, P.J.; John, C.S.; Nicholson, M.L.; Chapman, C.D.; Loera, E. Hypothalamic paraventricular 5-hydroxytryptamine inhibits the effects of ghrelin on eating and energy substrate utilization. Pharmacol. Biochem. Behav. 2010, 97, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Recinella, L.; Orlando, G.; Michelotto, B.; di Nisio, C.; Vacca, M. Effects of ghrelin and amylin on dopamine, norepinephrine and serotonin release in the hypothalamus. Eur. J. Pharmacol. 2002, 454, 189–192. [Google Scholar] [CrossRef]
- Pandit, R.; Omrani, A.; Luijendijk, M.C.M.; de Vrind, V.A.J.; van Rozen, A.J.; Ophuis, R.J.A.O.; Garner, K.; Kallo, I.; Ghanem, A.; Liposits, Z.; et al. Melanocortin 3 receptor signaling in midbrain dopamine neurons increases the motivation for food reward. Neuropsychopharmacology 2016, 41, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- Roselli-Rehfuss, L.; Mountjoy, K.G.; Robbins, L.S.; Mortrud, M.T.; Low, M.J.; Tatro, J.B.; Entwistle, M.L.; Simerly, R.B.; Cone, R.D. Identification of a receptor for γ melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc. Natl. Acad. Sci. USA 1993, 90, 8856–8860. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.M.; Irani, B.G.; Moore, M.C.; Haskell-Luevano, C.; Millard, W.J. Ghrelin-induced food intake and growth hormone secretion are altered in melanocortin 3 and 4 receptor knockout mice. Peptides 2005, 26, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.K.; Chow, K.B.; Lau, P.N.; Chu, K.M.; Chan, C.B.; Cheung, C.H.; Wise, H. The truncated ghrelin receptor polypeptide (GHSR-1b) acts as a dominant-negative mutant of the ghrelin receptor. Cell Signal. 2007, 19, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Jerlhag, E.; Egecioglu, E.; Dickson, S.L.; Engel, J.A. Ghrelin receptor antagonism attenuates cocaine- and amphetamine-induced locomotor stimulation, accumbal dopamine release, and conditioned place preference. Psychopharmacology 2010, 211, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [PubMed]
- Vucetic, Z.; Reyes, T.M. Central dopaminergic circuitry controlling food intake and reward: Implications for the regulation of obesity. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Wellman, P.J.; Davis, K.W.; Nation, J.R. Augmentation of cocaine hyperactivity in rats by systemic ghrelin. Regul. Pept. 2005, 125, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Enjalbert, A.; Bockaert, J. Pharmacological characterization of the D2 dopamine receptor negatively coupled with adenylate cyclase in rat anterior pituitary. Mol. Pharmacol. 1983, 23, 576–584. [Google Scholar] [PubMed]
- Kebabian, J.W.; Calne, D.B. Multiple receptors for dopamine. Nature 1979, 277, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Kebabian, J.W.; Greengard, P. Dopamine-sensitive adenyl cyclase: Possible role in synaptic transmission. Science 1971, 174, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Spano, P.F.; Govoni, S.; Trabucchi, M. Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem. Psychopharmacol. 1978, 19, 155–165. [Google Scholar] [PubMed]
- Fetissov, S.; Meguid, M.M.; Sato, T.; Zhang, L. Expression of dopaminergic receptors in the hypothalamus of lean and obese zucker rats and food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R905–R910. [Google Scholar] [CrossRef] [PubMed]
- Suchankova, P.; Engel, J.A.; Jerlhag, E. Sub-chronic ghrelin receptor blockade attenuates alcohol- and amphetamine-induced locomotor stimulation in mice. Alcohol Alcohol. 2015, 51, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.J.; Al-Naser, H.A.; Clifton, P.G. The anorectic effect of the selective dopamine D1-receptor agonist A-77636 determined by meal pattern analysis in free-feeding rats. Eur. J. Pharmacol. 2006, 532, 253–257. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, E.C.; Kremer, Y.; Lefort, S.; Harada, M.; Pascoli, V.; Rohner, C.; Lüscher, C. Accumbal D1R neurons projecting to lateral hypothalamus authorize feeding. Neuron 2015, 88, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Terry, P.; Katz, J.L. Differential antagonism of the effects of dopamine D1-receptor agonists on feeding behavior in the rat. Psychopharmacology 1992, 109, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.H.; Temple, J.L.; Neaderhiser, B.J.; Salis, R.J.; Erbe, R.W.; Leddy, J.J. Food reinforcement, the dopamine D2 receptor genotype, and energy intake in obese and nonobese humans. Behav. Neurosci. 2007, 121, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Pijl, H. Reduced dopaminergic tone in hypothalamic neural circuits: Expression of a “thrifty” genotype underlying the metabolic syndrome? Eur. J. Pharmacol. 2003, 480, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, C.P.; de Freitas, M.F.L.; Nogueira, M.I.; Campina, R.C.D.F.; Takase, L.F.; de Souza, S.L.; de Castro, R.M. Modulatory role of serotonin on feeding behavior. Nutr. Neurosci. 2010, 13, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Saller, C.F.; Stricker, E.M. Hyperphagia and increased growth in rats after intraventricular injection of 5,7-dihydroxytryptamine. Science 1976, 192, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Meguid, M.M.; Fetissov, S.O.; Varma, M.; Sato, T.; Zhang, L.; Laviano, A.; Rossi-Fanelli, F. Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition 2000, 16, 843–857. [Google Scholar] [CrossRef]
- Clifton, P.G.; Lee, M.D.; Dourish, C.T. Similarities in the action of Ro 60–0175, a 5-HT2C receptor agonist, and d-fenfluramine on feeding patterns in the rat. Psychopharmacology 2000, 152, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.C.; Barnes, N.M. Anti-obesity pharmacotherapy: Future perspectives utilising 5-HT2C receptor agonists. Drug Discov. Today Ther. Strateg. 2006, 3, 577–583. [Google Scholar] [CrossRef]
- Kennett, G.A.; Wood, M.D.; Bright, F.; Trail, B.; Riley, G.; Holland, V.; Avenell, K.Y.; Stean, T.; Upton, N.; Bromidge, S.; et al. SE 242084, a selective and brain penetrant 5-HT2C receptor antagonist. Neuropharmacology 1997, 36, 609–620. [Google Scholar] [CrossRef]
- Lam, D.D.; Przydzial, M.J.; Ridley, S.H.; Yeo, G.S.H.; Rochford, J.J.; O’Rahilly, S.; Heisler, L.K. Serotonin 5-HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology 2008, 149, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; de Vry, J. Role of 5-HT2C receptors in the hypophagic effect of m-CPP, ORG 37684 and CP-94,253 in the rat. Prog. Neuropsychopharmacol. Biol. Psychiatry 2002, 26, 441–449. [Google Scholar]
- Tecott, L.H.; Sun, L.M.; Akana, S.F.; Strack, A.M.; Lowenstein, D.H.; Dallman, M.F.; Julius, D. Eating disorder and epilepsy in mice lacking 5-HT2C Serotonin Receptors. Nature 1995, 374, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Tecott, L.H.; Abdallah, L. Mouse genetic approaches to feeding regulation: Serotonin 5-HT2C receptor mutant mice. CNS Spectr. 2003, 8, 578–588. [Google Scholar] [CrossRef]
- Millan, M.J.; Marin, P.; Bockaert, J.; Mannoury la Cour, C. Signaling at G-protein-coupled serotonin receptors: Recent advances and future research directions. Trends Pharmacol. Sci. 2008, 29, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Kesterson, R.A.; Khong, K.; Cullen, M.J.; Pelleymounter, M.A.; Dekoning, J.; Baetscher, M.; Cone, R.D. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 2000, 141, 3518–3521. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.S.; Marsh, D.J.; Trumbauer, M.E.; Frazier, E.G.; Guan, X.M.; Yu, H.; Rosenblum, C.I.; Vongs, A.; Feng, Y.; Cao, L.; et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 2000, 26, 97–102. [Google Scholar] [PubMed]
- Sutton, G.M.; Perez-Tilve, D.; Nogueiras, R.; Fang, J.; Kim, J.K.; Cone, R.D.; Gimble, J.M.; Tschop, M.H.; Butler, A.A. The melanocortin-3 receptor is required for entrainment to meal intake. J. Neurosci. 2008, 28, 12946–12955. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Pronchuk, N.; Fan, W.; Dinulescu, D.M.; Colmers, W.F.; Cone, R.D. Integration of NPY, AgRP, and melanocortin signals in the hypothalamic paraventricular nucleus: Evidence of a cellular basis for the adipostat. Neuron 1999, 24, 155–163. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Boston, B.A.; Kesterson, R.A.; Hruby, V.J.; Cone, R.D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997, 385, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, A.; Conwell, I.M.; Hruby, V.; Mayorov, A.; Cai, M.; Wardlaw, S.L. Effects of selective modulation of the central melanocortin-3-receptor on food intake and hypothalamic POMC expression. Peptides 2008, 29, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.L.; Hruby, V.; Brookhart, G.; Cone, R.D. The regulation of food intake by selective stimulation of the type 3 melanocortin receptor (MC3R). Peptides 2006, 27, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Bagnol, D.; Lu, X.Y.; Kaelin, C.B.; Day, H.E.; Ollmann, M.; Gantz, I.; Akil, H.; Barsh, G.S.; Watson, S.J. Anatomy of an endogenous antagonist: Relationship between agouti-related protein and proopiomelanocortin in brain. J. Neurosci. 1999, 19, 1–7. [Google Scholar]
- Jégou, S.; Boutelet, I.; Vaudry, H. Melanocortin-3 receptor mRNA expression in pro-opiomelanocortin neurones of the rat arcuate nucleus. J. Neuroendocrinol. 2000, 12, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Fong, T.M. The melanocortin system. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E468–E474. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
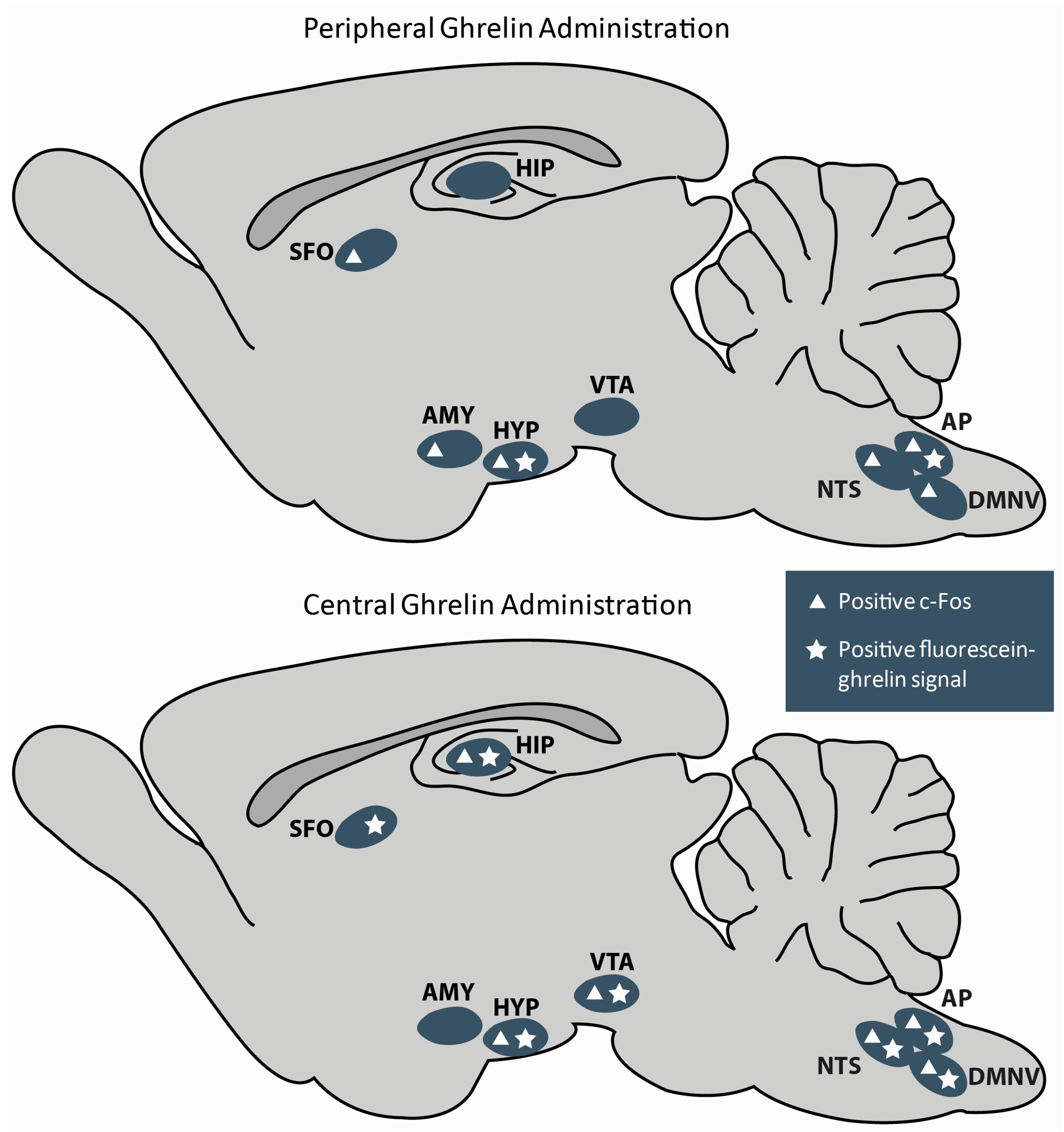{kind=link}
| Technique(s) | Findings Summary | Species | Reference(s) | |
|---|---|---|---|---|
| Supporting Evidence | RT PCR | Ghrelin transcript detected in the brain | Rat | [20] |
| RT PCR | Ghrelin transcript detected in the arcuate nucleus (ARC) | Rat | [24] | |
| RT PCR | Ghrelin transcript detected in the hypothalamus (HYP) of wild-type (WT) but not ghrelin knockouts (KOs) | Mouse | [22] | |
| RT PCR | Ghrelin mRNA detected in HYP | Rat | [215] | |
| RT PCR | Ghrelin mRNA detected in cerebral cortex and HYP | Rat | [214] | |
| RT PCR | Hypothalamic expression of ghrelin significantly lower in clock mutants | Mouse | [217] | |
| RT PCR | Hypothalamic expression of ghrelin and ghrelin O-acyltransferase (GOAT) mRNA increases following a 48 h fast | Rat | [56] | |
| RT PCR and transgenics | Detected ghrelin and enhanced green fluorescent protein (EGFP) transcripts in the HYP of transgenic mice that were engineered to express EGFP in cells that produce ghrelin (i.e., inserted the gene for EGFP in the regulatory region of the ghrelin gene) | Mouse | [225] | |
| Immunohistochemistry | Ghrelin-immunoreactive neurons detected in the ARC | Rat | [20,24,220,221,223] | |
| Immunohistochemistry | Numerous ghrelin immunoreactive cell bodies and axons found within the HYP;small number of ghrelin immunoreactive processes found within extrahypothalamic brain regions (e.g., BNST, NA, cortex) | Mouse | [22] | |
| Immunohistochemistry | Detected ghrelin immunoreactivity in the HYP | Human | [224] | |
| Immunohistochemistry | Ghrelin immunoreactive neurons were detected in the HYP and cerebral cortex | Rat | [214] | |
| Immunohistochemistry | HYP ghrelin was detected in sham operated rats but not in those that underwent a pinealectomy | Rat | [220] | |
| Immunohistochemistry and transgenics | EGFP fluorescence detected in ARC of transgenic ghrelin reporter mice (inserted the gene for EGFP in the regulatory region of the ghrelin gene) | Mouse | [225] | |
| Reverse phase high performance liquid chromatography and ghrelin radioimmuneassays | Detected ghrelin radioimmunoreactivity in the ARC | Rat | [24] | |
| Reverse phase high performance liquid chromatogra-phy and ghrelin radioimmuneassays | Detected ghrelin radioimmunoreactivity in the HYP | Rat | [215] | |
| Behavioural tests | Chronic central GOAT knockdown (via ICV infused amorpholino antisense oligonucleotides) significantly decreased weight gain of rats fed a high fat diet | Rat | [56] | |
| Refuting Evidence | RT PCR and transgenics | Failed to detect ghrelin immunoreactivity or ghrelin mRNA (via in situ hybridization) within the brain of WT or ghrelin-hrGFP BAC transgenic reporter mice | Mouse | [25] |
| Immunohisto-chemistry | Strong ghrelin immunoreactive signals from the stomach but no specific immunoreactivity for either ghrelin or des-acyl ghrelin in the central nervous system (e.g., HYP, medulla oblongata, and spinal cord) using four separate well characterized anti-ghrelin antibodies | Mouse and Rat | [23] | |
| Immunohisto-chemistry and transgenics | Evident β-galactosidase staining within the stomach and intestine but no positive staining within the HYP of transgenic ghrelin deficient mice (ghrl−/−) engineered to express a lacZ reporter gene in the place of the functional ghrelin gene WT and ghrelin deficient mice show negligible and indistinguishable hypothalamic ghrelin immunoreactivity despite having very different stomach ghrelin immunoreactivity profiles | Mouse | [218] | |
| Immunohisto-chemistry and transgenics | Failed to detect GFP fluorescence in the brain but demonstrated clear GFP expression in the stomach using two separate transgenic ghrelin reporter mice (i.e., ghrelin-hrGFP BAC transgenic mice), in which humanized Renilla reniformis GFP expression was driven by different lengths of the ghrelin promotor | Mouse | [25] |
| Heterodimer | In Vitro Observations of Heterodimer | Ex Vivo Observations of Heterodimer | Feeding Related Brain Regions Where Receptor Expression Overlaps | Known Cross Talk Between Systems | Influence of Heterodimer Over Feeding Behaviours |
|---|---|---|---|---|---|
| GHSR-1a/GHSR-1b | Low ratio of GHSR-1b relative to GHSR-1a ↑cell surface expression of the heterodimer and↑Gαi/o dependent signaling cascades following ghrelin administration [258] High ratio of GHSR-1b relative to GHSR-1a↓cell surface expression of the heterodimer and↓Gαi/o dependent signaling cascades following ghrelin administration [258] | Increasing the expression of GHSR-1b in striatal (i.e., naturally high GHSR-1b to GHSR-1a expression ratio) cultures↓ghrelin signalling efficiency [258] Increasing the expression of GHSR-1b in hippocampal (very low inherent GHSR-1b/GHSR-1a expression ratio) cultures↑ghrelin signalling efficiency [258] | HIP, NA [12,13,14,259] | If GHSR-1b subunit is much higher than GHSR-1a then the GHSR-1a/GHSR-1b heterodimer can still bind agonists but forfeits the capacity to stimulate associated signaling proteins [257] | Unknown but speculated↑in feeding when ratio of GHSR-1a to GHSR-1b is high but↓in feeding when ratio of GHSR-1a to GHSR-1b is low |
| GHSR-1a/DRD1 | Co-localize, immuno-precipitate together, and give off a reliable positive bioluminescence resonance energy transfer signal only in the presence of agonists (i.e., dopamine and ghrelin) [234] Enhances Ca2+ and cAMP accumulation in HEK cells in response to ghrelin and dopamine above those observed when GHSR or DR1D are expressed alone [234] | DRD1 agonists significantly enhance the formation of GHSR/DRD1 heterodimers within hippocampal neurons [157] DRD1 and GHSR agonists enhance Ca2+ in WT neurons that express the heterodimer but are unable to elicit this effect in GHSR antagonist treated and GHSR KO hippocampal neurons [157] | HIP, VTA, Striatum, Cortex [12,13,14,235] | Pre-treatment with a D1-like antagonist into the NA, completely blocks the rewarding effects of intra-VTA ghrelin [116] GHSR inactivation completely attenuates DRD1 regulated hippocampal behaviour and memory [256] | Unknown |
| GHSR-1a/DRD2 | DRD2 agonists↑intracellular calcium levels in HEK 293 cells which co-express both GHSR-1as and DRD2s but not in cells transfected with either receptor independently [235] HEK 293 cells that are pre-treated with either DRD2 or GHSR antagonists do not enhance intracellular calcium levels in response to DRD2 or GHSR agonists [235] | FRET detection of GHSR-1a/DRD2 heterodimers in hypothalamic preparations of WT but not GHSR KO mice [235] | HIP, HYP, Striatum [12,13,14,236] | DRD2 agonists known to suppress appetite, reliably↓food intake in WT but not GHSR KO mice [235] Pre-treatment of WT mice with GHSR antagonists, prevent decreases in feeding following cabergoline injections [235] Pre-treatment with a D2-like antagonist into the NA, completely blocks the rewarding effects of intra-VTA ghrelin [116] | Unknown but speculated attenuation of feeding behaviours |
| GHSR-1a/5-HT2C | When GHSR-1as and 5-HT2C receptors are co-transfected into HEK 293 cells they co-localize [236] GHSR-1a agonists significantly↑the co-internalization of these receptors in HEK 293 cells [236] Co-transfection of GHSR-1a and 5-HT2C in HEK 293 cells reduces GHSR agonist induced GHSR signaling activity (65% in Ca2+ mobilization) [236] | Unknown | HIP, HYP, VTA, Cortex [12,13,14,271,272,273] | Peripheral administration of 5-HT2C receptor agonists block the↑in plasma ghrelin seen following a 24 h fast and increases expression of anorexigenic peptides within the HYP [273] Intra-PVN microinjections of 5-HT2C agonists significantly attenuates the orexigenic effect of intra-PVN ghrelin [274,275] GHSR activation by ghrelin blocks the release of serotonin in hypothalamic synaptosomes [276] | Unknown but speculated attenuation of feeding behaviours |
| GHSR-1a/MC3-R | Co-transfection of GHSRs and MC3-Rs in COS-7 cells potentiates α-MSH induced cAMP accumulation [260] Co-transfection of these receptors in HEK 293 cells↓basal as well as ghrelin induced canonical GHSR signaling (i.e., >50%↓) [260] | Unknown | HYP, VTA [12,13,14,265,266,277,278] | The orexigenic effects of peripheral ghrelin in WT mice is lost in MC3-R KO mice [279] | Unknown but speculated↑in feeding |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, A.; Abizaid, A. Clarifying the Ghrelin System’s Ability to Regulate Feeding Behaviours Despite Enigmatic Spatial Separation of the GHSR and Its Endogenous Ligand. Int. J. Mol. Sci. 2017, 18, 859. https://doi.org/10.3390/ijms18040859
Edwards A, Abizaid A. Clarifying the Ghrelin System’s Ability to Regulate Feeding Behaviours Despite Enigmatic Spatial Separation of the GHSR and Its Endogenous Ligand. International Journal of Molecular Sciences. 2017; 18(4):859. https://doi.org/10.3390/ijms18040859
Chicago/Turabian StyleEdwards, Alexander, and Alfonso Abizaid. 2017. "Clarifying the Ghrelin System’s Ability to Regulate Feeding Behaviours Despite Enigmatic Spatial Separation of the GHSR and Its Endogenous Ligand" International Journal of Molecular Sciences 18, no. 4: 859. https://doi.org/10.3390/ijms18040859
APA StyleEdwards, A., & Abizaid, A. (2017). Clarifying the Ghrelin System’s Ability to Regulate Feeding Behaviours Despite Enigmatic Spatial Separation of the GHSR and Its Endogenous Ligand. International Journal of Molecular Sciences, 18(4), 859. https://doi.org/10.3390/ijms18040859