
New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease


Abstract
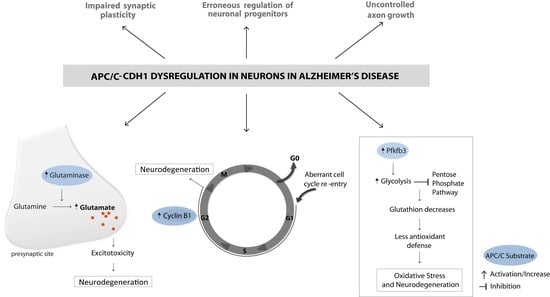
:
1. Introduction
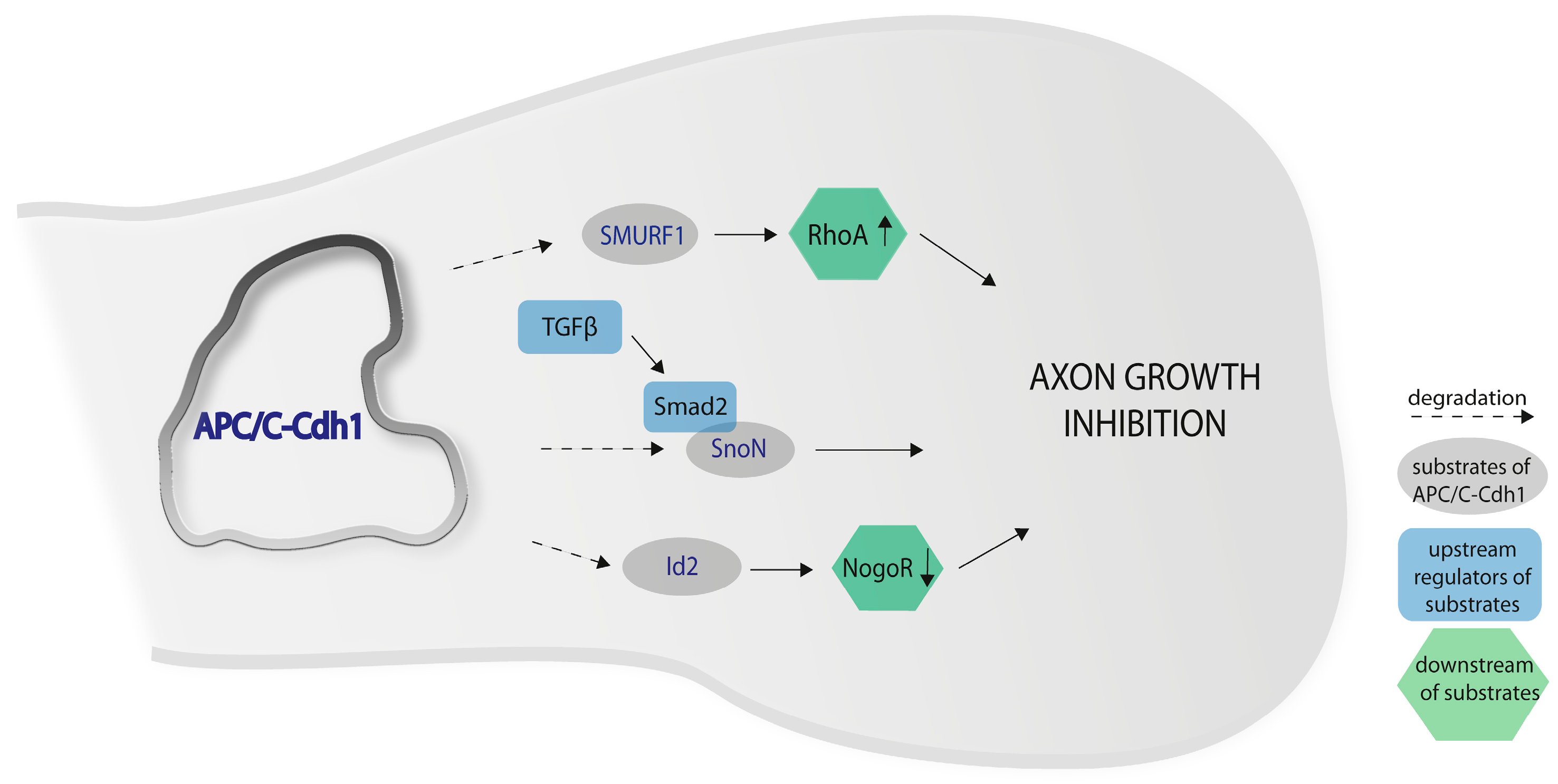
2. APC/C-Cdh1 and Axon Growth in the Cerebellar Cortex
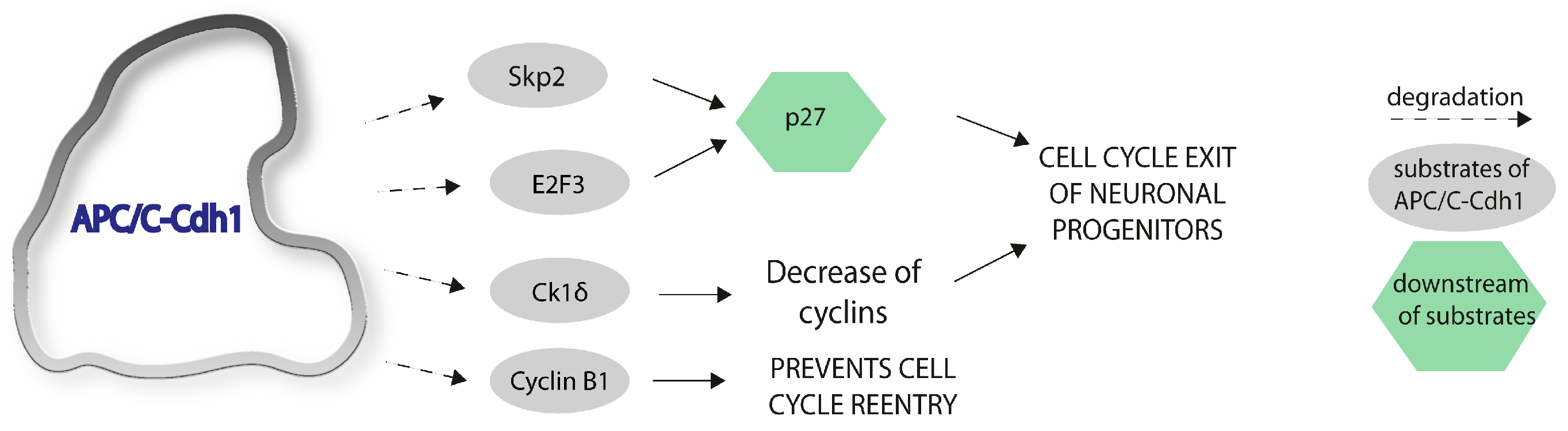
3. APC/C-Cdh1 and Neurogenesis
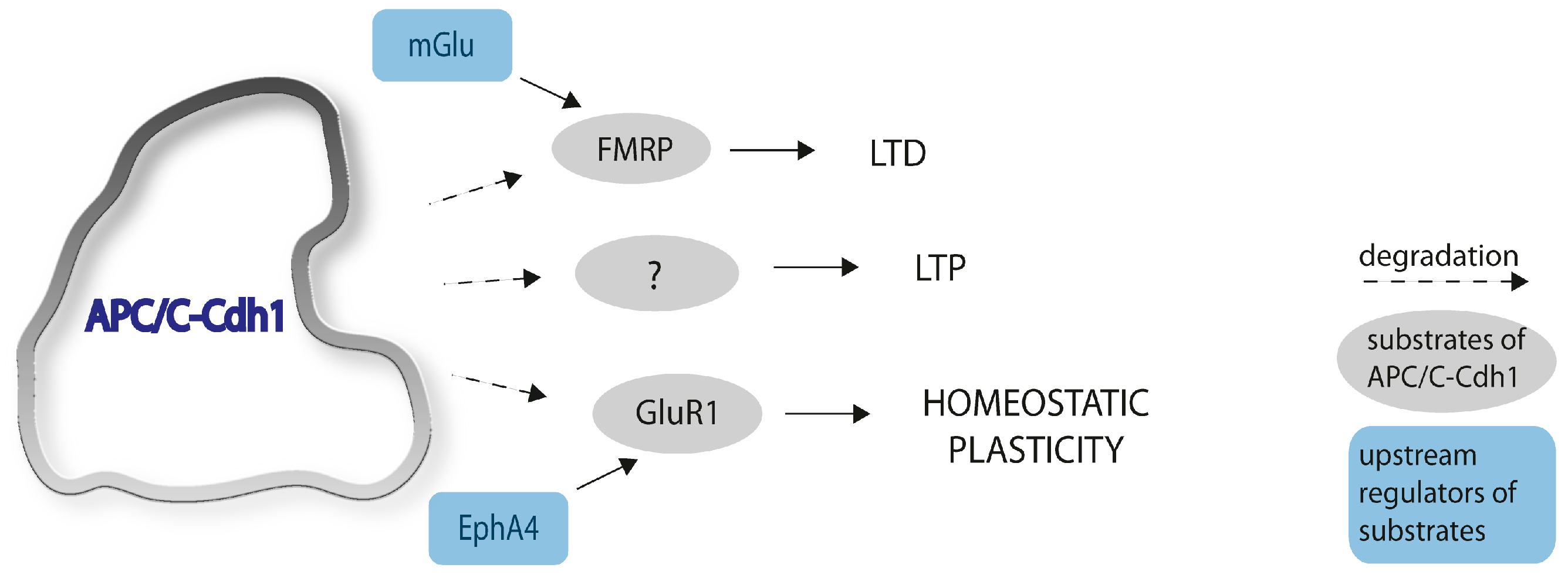
4. APC/C-Cdh1 Is Involved in Synaptic Plasticity
4.1. APC/C-Cdh1 and Long-Term Potentiation
4.2. APC/C-Cdh1 and Long-Term Depression
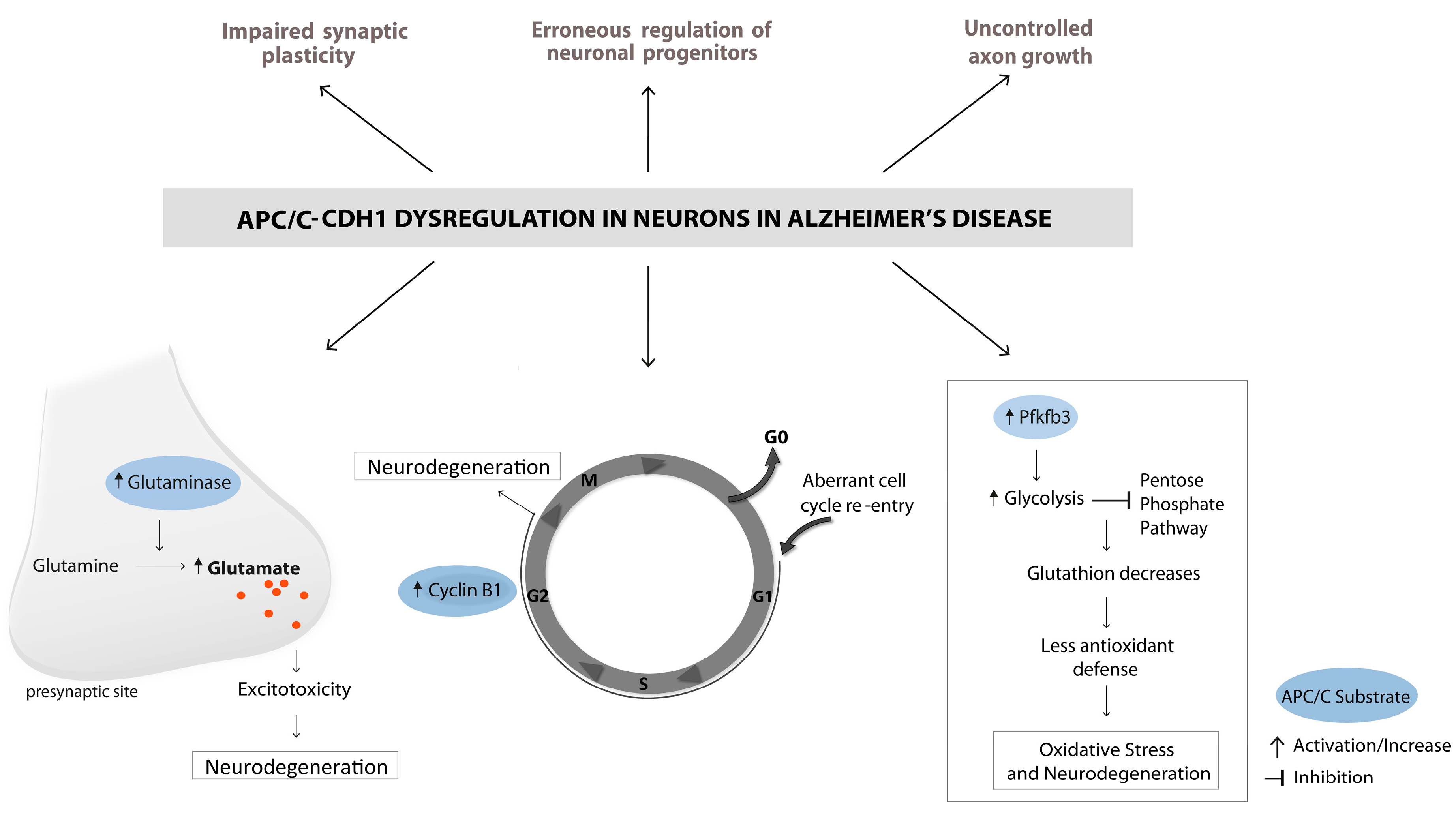
5. APC/C and Its Emerging Role in Alzheimer’s Disease
6. The Ectopic Cell Cycle in AD and Its Relation to APC/C-Cdh1
7. Oxidative Stress in AD and Its Relation to APC/C-Cdh1
8. Excitotoxicity in AD and Its Relation to APC/C-Cdh1
9. LTP Impairment in AD and Its Relation to APC/C-Cdh1
10. Impaired Neurogenesis in AD and Its Relation to APC/C-Cdh1
11. Conclusions
Acknowledgments
Conflicts of Interest
References
- Tomoko, Y.; Yang, Y.; Bonni, A. Spatial Organization of Ubiquitin Ligase Pathways Orchestrates Neuronal Connectivity. Trends Neurosci. 2013, 36, 218–226. [Google Scholar]
- King, R.W.; Glotzer, M.; Kirschner, M.W. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol. Biol. Cell 1996, 7, 1343–1357. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.F.; Zhang, Z.; Yang, J.; McLaughlin, S.H.; Barford, D. Molecular architecture and mechanism of the anaphase-promoting complex. Nature 2014, 51, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Visintin, R.; Prinz, S.; Amon, A. CDC20 and CDH1: A family of substrate-specific activators of APC-dependent proteolysis. Science 1997, 278, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, M.; Murray, A.W.; Kirschner, M.W. Cyclin is degraded by the ubiquitin pathway. Nature 1991, 349, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, C.M.; Kirschner, M.W. The KEN box: An APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000, 14, 655–665. [Google Scholar] [PubMed]
- Reis, A.; Levasseur, M.; Chang, H.Y.; Elliott, D.J.; Jones, K.T. The CRY box: A second APC cdh1-dependent degron in mammalian cdc20. EMBO Rep. 2006, 7, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Zhang, L.; Gamper, A.M.; Fujita, T.; Wan, Y. APC/C-Cdh1: From cell cycle to cellular differentiation and genomic integrity. Cell Cycle 2010, 9, 3904–3912. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Bolaños, J.P.; Moncada, S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc. Natl. Acad. Sci. USA 2010, 107, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Esteban, M.; García-Higuera, I.; Maestre, C.; Moreno, S.; Almeida, A. APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat. Commun. 2013, 4, 2879. [Google Scholar] [CrossRef] [PubMed]
- Gieffers, C.; Peters, B.H.; Kramer, E.R.; Dotti, C.G.; Peters, J.M. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 11317–11322. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Stegmuller, J.; Matsuda, T.; Bonni, S.; Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 2004, 303, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Stegmüller, J.; Konishi, Y.; Huynh, M.A.; Yuan, Z.; Dibacco, S.; Bonni, A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron 2006, 50, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Stegmüller, J.; Guardavaccaro, D.; Liu, G.; Carro, M.S.; Rothschild, G.; de la Torre-Ubieta, L.; Pagano, M.; Bonni, A.; Iavarone, A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 2006, 442, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Harmey, D.; Smith, A.; Simanski, S.; Moussa, Z.C.; Ayad, N.G. The Anaphase Promoting Complex Induces Substrate Degradation during Neuronal Differentiation. J. Biol. Chem. 2009, 284, 4317–4323. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Hung, K.W.; Fu, W.Y.; Shen, C.; Chen, Y.; Xia, J.; Lai, K.O.; Ip, N.Y. APC Cdh1 mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat. Neurosci. 2011, 14, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shin, Y.H.; Hou, L.; Huang, X.; Wie, Z.; Klann, E.; Zhang, P. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat. Cell Biol. 2008, 10, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ikeuchi, Y.; Malumbres, M.; Bonni, A. A Cdh1-APC/FMRP Ubiquitin Signaling Link Drives mGluR-Dependent Synaptic Plasticity in the Mammalian Brain. Neuron 2015, 86, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Puram, S.V.; Bilimoria, P.M.; Ikeuchi, Y.; Keough, S.; Wong, M.; Rowitch, D.; Bonni, A. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 2009, 136, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kim, A.H.; Yamada, T.; Wu, B.; Bilimoria, P.M.; Ikeuchi, Y.; de la Iglesia, N.; Shen, J.; Bonni, A. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 2009, 326, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, T.; Martínez-Bellver, S.; Giraldo, E.; Teruel-Martí, V.; Lloret, A.; Viña, J. Aβ Induces Excitotoxicity Mediated by APC/C-Cdh1 Depletion That Can Be Prevented by Glutaminase Inhibition Promoting Neuronal Survival. Sci. Rep. 2016, 6, 31158. [Google Scholar] [CrossRef] [PubMed]
- Maestre, C.; Delgado-Esteban, M.; Gomez-Sanchez, J.C.; Bolaños, J.P.; Almeida, A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008, 20, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, P.; Fernandez, E.; Almeida, A.; Bolanos, J.P. Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ. 2012, 10, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Clark, A.W.; Rosales, J.L.; Chapman, K.; Fung, T.; Johnston, R. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci. Res. 1999, 34, 21–29. [Google Scholar] [CrossRef]
- Tseng, H.C.; Zhou, Y.; Shen, Y.; Tsai, L.H.A. Survey of Cdk5 activator p35 and p25 levels in Alzheimer’s disease brains. FEBS Lett. 2002, 523, 58–62. [Google Scholar] [CrossRef]
- Zhang, J.; Cicero, S.A.; Wang, L.; Romito-DiGiacomo, R.R.; Yang, Y.; Herrup, K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 8772–8777. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative damage in Alzheimer’s. Nature 1996, 382, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Van Roessel, P.; Elliott, D.A.; Robinson, I.M.; Prokop, A.; Brand, A.H. Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell 2004, 119, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, J.R.; Dube, H.; Touroutine, D.; Rush, K.M.; Goodwin, P.R.; Carozza, M.; Didier, Z.; Francis, M.M.; Juo, P. The Anaphase-Promoting Complex (APC) ubiquitin ligase regulates GABA transmission at the C. elegans neuromuscular junction. Mol. Cell. Neurosci. 2014, 58, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Stroschein, S.L.; Bonni, S.; Wrana, J.L.; Luo, K. Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 2001, 15, 2822–2836. [Google Scholar] [PubMed]
- Kannan, M.; Lee, S.J.; Schwedhelm-Domeyer, N.; Stegmüller, J. The E3 ligase Cdh1-anaphase promoting complex operates upstream of the E3 ligase Smurf1 in the control of axon growth. Development 2012, 139, 3600–3612. [Google Scholar] [CrossRef] [PubMed]
- Penas, C.; Govek, E.E.; Fang, Y.; Ramachandran, V.; Daniel, M.; Wang, W.; Maloof, M.E.; Rahaim, R.J.; Bibian, M.; Kawauchi, D.; et al. Casein Kinase 1d Is an APC/CCdh1 Substrate that Regulates Cerebellar Granule Cell Neurogenesis. Cell Rep. 2015, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Lim, R.; Bashir, T.; Pagano, M.; Guardavaccaro, D. APC/CCdh1 controls the proteasome-mediated degradation of E2F3 during cell cycle exit. Cell Cycle 2012, 11, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.L.; Palacios-Callender, M.; Frakich, N.; de Leon, J.; Schmitt, C.A.; Boorn, L.; Davis, N.; Moncada, S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 18868–18873. [Google Scholar] [CrossRef] [PubMed]
- García-Higuera, I.; Manchado, E.; Dubus, P.; Cañamero, M.; Méndez, J.; Moreno, S.; Malumbres, M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008, 10, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Hainline, S.G.; Rickmyre, J.L.; Neitzel, L.R.; Lee, L.A.; Lee, E. The Drosophila MCPH1-B isoform is a substrate of the APCCdh1 E3 ubiquitin ligase complex. Biol. Open 2014, 3, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.L.; Lu, H.; Shelly, M.; Gao, H.; Poo, M.M. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 2011, 69, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Heuckeroth, R.O. Retinoic acid regulates murine enteric nervous system precursor proliferation, enhances neuronal precursor differentiation, and reduces neurite growth in vitro. Dev. Biol. 2008, 320, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Bashir, T.; Dorrello, N.V.; Amador, V.; Guardavaccaro, D.; Pagano, M. Control of the SCF (Skp2-Cks1) ubiquitin ligase by the APC/C (Cdh1) ubiquitin ligase. Nature 2004, 428, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Eguren, M.; Porlan, E.; Manchado, E.; García-Higuera, I.; Cañamero, M.; Fariñas, I.; Malumbres, M. The APC/C cofactor Cdh1 prevents replicative stress and p53-dependent cell death in neural progenitors. Nat. Commun. 2013, 4, 2880. [Google Scholar] [CrossRef] [PubMed]
- Juo, P.; Kaplan, J.M. The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Curr. Biol. 2004, 14, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Pick, J.E.; Wang, L.; Mayfield, J.E.; Klann, E. Neuronal Expression of the Ubiquitin E3 ligase APC/C-Cdh1 during Development is Required for Long-term Potentiation, Behavioral Flexibility, and Extinction. Neurobiol. Learn. Mem. 2013, 100, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Pick, J.E.; Malumbres, M.; Klann, E. The E3 ligase APC/C-Cdh1 is required for associative fear memory and long-term potentiation in the amygdala of adult mice. Learn. Mem. 2013, 20, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Khodosevich, K.; Monyer, H. Dendrite development regulated by the schizophrenia-associated gene FEZ1 involves the ubiquitin proteasome system. Cell Rep. 2014, 24, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A. Regulation of APC/C-Cdh1 and Its Function in Neuronal Survival. Mol. Neurobiol. 2012, 46, 547–554. [Google Scholar] [CrossRef] [PubMed]
- McShea, A.; Harris, P.L.; Webster, K.R.; Wahl, A.F.; Smith, M.A. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 1933–1939. [Google Scholar] [PubMed]
- Nagy, Z.; Esiri, M.M.; Smith, A.D. Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol. 1997, 93, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.W.; Lippa, C.F. Ki-67 immunoreactivity in Alzheimer’s disease and other neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 1995, 54, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Evans, T.A.; Santocanale, C.; Llosá, J.C.; Viña, J.; Bajic, V.; Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A.; et al. Evidence for the progression through S-phase in the ectopic cell cycle reentry of neurons in Alzheimer disease. Aging 2009, 1, 382–388. [Google Scholar]
- Copani, A.; Caraci, F.; Hoozemans, J.M.; Calafiore, M.; Sortino, M.A.; Nicoletti, F. The nature of the cell cycle in neurons: Focus on a “non-caconical” pathway of DNA replication causally related to death. Biochim. Biophys. Acta 2007, 772, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.; Jicha, G.; Rosado, M.; Dickson, D.W. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J. Neurosci. 1997, 17, 3588–3598. [Google Scholar] [PubMed]
- Lopes, J.P.; Agostinho, P. Cdk5: Multitasking between physiological and pathological conditions. Prog. Neurobiol. 2011, 94, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, H.; Zhou, T.; Zhou, J.; Herrup, K. Cdk5 levels oscillate during the neuronal cell cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. J. Biol. Chem. 2012, 287, 25985–25994. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badía, M.C.; Mora, N.J.; Ortega, A.; Pallardó, F.V.; Alonso, M.D.; Atamna, H.; Viña, J. Gender and age-dependent differences in the mitochondrial apoptogenic pathway in Alzheimer’s disease. Free Radic. Biol. Med. 2008, 44, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Shi, D.; Westaway, D.; Jhamandas, J.H. Bioenergetic mechanisms in astrocytes may contribute to amyloid plaque deposition and toxicity. J. Biol. Chem. 2015, 290, 12504–12513. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s Disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Pomara, N.; Singh, R.; Deptula, D.; Chou, J.C.; Schwartz, M.B.; LeWitt, P.A. Glutamate and other CSF amino acids in Alzheimer’s disease. Am. J. Psychiatry 1992, 149, 251–254. [Google Scholar] [PubMed]
- Csernansky, J.G.; Bardgett, M.E.; Sheline, Y.I.; Morris, J.C.; Olney, J.W. CSF excitatory amino acids and severity of illness in Alzheimer’s disease. Neurology 1996, 46, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Molina, J.A.; Gómez, P.; Vargas, C.; de Bustos, F.; Benito-León, J.; Tallón-Barranco, A.; Ortí-Pareja, M.; Gasalla, T.; Arenas, J. Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. J. Neural Transm. 1998, 105, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Schoenknecht, P.; Kassner, S.; Hildebrandt, W.; Kinscherf, R.; Schroeder, J. Cerebrospinal fluid concentrations of functionally important amino acids and metabolic compounds in patients with mild cognitive impairment and Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Toldi, J.; Vécsei, L. Glutamatergic dysfunctioning in Alzheimer’s disease and related therapeutic targets. J. Alzheimers Dis. 2014, 42, 177–187. [Google Scholar]
- Lipton, S.A. The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: Low-affinity, uncompetitive antagonism. Curr. Alzheimer Res. 2005, 2, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Lauderback, C.M.; Harris-White, M.E.; Wang, Y.; Pedigo, N.W.; Carney, J.M.; Butterfield, D.A. Amyloid β-peptide inhibits Na+-dependent glutamate uptake. Life Sci. 1999, 65, 1977–1981. [Google Scholar] [CrossRef]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β Slows Clearance of Synaptically Released Glutamate by Mislocalizing Astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; McGeer, P.L.; Itagaki, S.; McGeer, E.G.; Kaneko, T. Loss of glutaminase-positive cortical neurons in Alzheimer’s disease. Neurochem. Res. 1989, 14, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Kowall, N.W.; Beal, M.F. Glutamate-, glutaminase-, and taurine-immunoreactive neurons develop neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1991, 29, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Burbaeva, G.S.; Boksha, I.S.; Tereshkina, E.B.; Savushkina, O.K.; Starodubtseva, L.I.; Turishcheva, M.S. Glutamate metabolizing enzymes in prefrontal cortex of Alzheimer’s disease patients. Neurochem. Res. 2005, 30, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Tanaka, K.; Dawe, G.S.; Ittner, L.M.; Farooqui, A. Slow excitotoxicity in Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 643–668. [Google Scholar] [PubMed]
- Norris, C.M.; Blalock, E.M.; Thibault, O.; Brewer, L.D.; Clodfelter, G.V.; Porter, N.M.; Landfield, P.W. Electrophysiological mechanisms of delayed excitotoxicity: Positive feedback loop between NMDA receptor current and depolarization-mediated glutamate release. J. Neurophysiol. 2006, 96, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.S.; Wei, W.Z.; Shimahara, T.; Xie, C.W. Alzheimer amyloid β-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol. Learn. Mem. 2002, 77, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Di Lorenzo, F.; Bonnì, S.; Ponzo, V.; Caltagirone, C.; Martorana, A. Impaired LTP- but not LTD-like cortical plasticity in Alzheimer’s disease patients. J. Alzheimers Dis. 2012, 31, 593–599. [Google Scholar] [PubMed]
- Gengler, S.; Hamilton, A.; Hölscher, C. Synaptic plasticity in the hippocampus of a APP/PS1 mouse model of Alzheimer’s disease is impaired in old but not young mice. PLoS ONE 2010, 5, e9764. [Google Scholar] [CrossRef] [PubMed]
- Simón, A.M.; de Maturana, R.L.; Ricobaraza, A.; Escribano, L.; Schiapparelli, L.; Cuadrado-Tejedor, M.; Pérez-Mediavilla, A.; Avila, J.; Del Río, J.; Frechilla, D. Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; He, J.; Zhang, Y.; Luo, H.; Zhu, S.; Yang, Y.; Zhao, T.; Wu, J.; Huang, Y.; Kong, J.; et al. Increased hippocampal neurogenesis in the progressive stage of Alzheimer’s disease phenotype in an APP/PS1 double transgenic mouse model. Hippocampus 2009, 12, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yamamori, H.; Tatebayashi, Y.; Shafit-Zagardo, B.; Tanimukai, H.; Chen, S.; Iqbal, K.; Grundke-Iqbal, I. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 2008, 67, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer’s disease. Cell Death Dis. 2011, 2, e120. [Google Scholar] [CrossRef] [PubMed]
- Cedeño, C.; la Monaca, E.; Esposito, M.; Gutierrez, G.J. Detection and Analysis of Cell Cycle-Associated APC/C-Mediated Cellular Ubiquitylation in vitro and in vivo. Methods Mol. Biol. 2016, 1449, 251–265. [Google Scholar] [PubMed]
- Liu, Z.; Yuan, F.; Ren, J.; Cao, J.; Zhou, Y.; Yang, Q.; Xue, Y. GPS-ARM: Computational Analysis of the APC/C Recognition Motif by Predicting D-Boxes and KEN-Boxes. PLoS ONE 2012, 7, e34370. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt Entry/Organism | Gene | Protein (APC/C Substrate) | Function | Motif | Coactivator | Reference |
|---|---|---|---|---|---|---|
| Q63689 | Neurod2 | Neurogenic differentiation factor 2 | Regulation of presynaptic differentiation | D-box | cdc20 | [20] |
| Rattus norvegicus | ||||||
| Q9VM93 | syd-2 | Liprin-α | Regulation of synaptic size and activity/ γ-aminobutyric acid release at neuromuscular junction | D-box | cdh1/cdc20 | [28,29] |
| Caenorhabditis elegans | ||||||
| P19490 | Gria1 | Glutamate receptor 1 (GluR1) | Regulation of AMPA receptors in homeostatic plasticity | D-box | cdh1 | [16] |
| Rattus norvegicus | ||||||
| P41135 | Id1 | DNA-binding protein inhibitor ID-1 | Involved in dendrite morphogenesis in neurons | D-box | cdh1/cdc2 | [19] |
| Rattus norvegicus | ||||||
| Q60665 | Skil | Ski-like protein (SnoN) | Regulation of axonal morphogenesis | D-box | cdh1 | [13,30] |
| Mus musculus | ||||||
| Q9CUN6 | Smurf1 | E3 ubiquitin-protein ligase SMURF1 | E3 Ubiquitin ligase that targets RhoA, regulates axon growth | D-box | cdh1 | [31] |
| Mus musculus | ||||||
| P41137 | Id2 | DNA-binding protein inhibitor 2 ID-2 | Links axonal growth and cell cycle exit | D-box | cdh1 | [14] |
| Rattus norvegicus | ||||||
| Q06486 | Csnk1d | Casein kinase I isoform delta | Regulation neurogenesis in cerebellum | D-box | cdh1 | [32] |
| Rattus norvegicus | ||||||
| O00716 | E2F3 | Transcription factor E2F3 | Cell cycle exit and neuronal differentiation | D-box | cdh1 | [33] |
| Homo sapiens | ||||||
| P07818 | Ccnb1 | G2/mitotic-specific cyclin-B1 | Maintains cell cycle exit and promotes neuronal survival | D-box | cdh1 | [2,22] |
| Rattus norvegicus | ||||||
| P07953 | Pfkfb3 | 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) | Regulation of glycolytic pathway in neurons, oxidative stress | KEN box | cdh1 | [23,34] |
| Rattus norvegicus | ||||||
| P13264 | Gls | Glutaminase kidney isoform, mitochondrial | Regulates levels of neurotransmitter glutamate in neurons | KEN box | cdh1 | [21,35] |
| Rattus norvegicus | ||||||
| P35922 | Fmr1 | Synaptic functional regulator FMR1 (FMRP) | Drives mGluR-dependent synaptic plasticity | D-box | cdh1 | [18] |
| Mus musculus | ||||||
| Q8K0X8 | Fez1 | Fasciculation and elongation protein zeta-1 (FEZ1) | Dendrite growth in hippocampus | D-box | cdc20 | [36] |
| Mus musculus | ||||||
| Q8NEM0 | MCPH1 | Microcephalin MCPH1 (isoform B) | Cell cycle protein; homolog of a causative gene for autosomal recessive primary microcephaly in humans | D-box | cdh1 | [37] |
| Homo sapiens |
| Cdh1 knock-out (Fzr1 gene) | Description | Phenotype defetcs | Reference |
|---|---|---|---|
| Gene-trap (gt) construct, inserted into intron 5 of Fzr1, generating a dysfunctional allele of Fzr1 | Homozygous mice Cdh1gt/gt (intercrossed heterozygous mice Cdh1gt) | Early embryonic lethality (died at ~E9.5), replicative senescence, premature fibroblasts | [17] |
| Heterozygous Cdh1gt (50% cdh1 reduction) | Defects in hippocampal late phase LTP, deficiency in contextual fear-conditioning | [17] | |
| Two loxP sites eliminate exons 2 and 3 from the Frz1 gene, cre recombinase expressed under the Sox2 promoter | Conditional cdh1 knockout mice (embryo restricted knockout) | Loss of genomic stability, increased susceptibility to spontaneous tumours | [45] |
| Replicative stress, p53-mediated apoptotic death, alterations in neurogenesis resembling microcephaly | [10] | ||
| Two loxP sites eliminate exons 2 and 3 from the Frz1 gene, cre recombinase expressed under nestin regulatory sequences | Conditional cdh1 knockout mice (knockout restricted to the developing nervous system) | Hypoplastic brain and hydrocephalus | [41] |
| Two loxP sites eliminate exons 2 and 3 from the Frz1 gene, cre recombinase expressed under CaMKII promoter | Conditional cdh1 knockout mice (knockout restricted to excitatory neurons in the hippocampus and forebrain) | Impaired memory, impaired LTP in amygdala | [44] |
| Two loxP sites eliminate exons 2 and 3 from the Frz1 gene, cre recombinase exressed under enolase promoter | Conditional cdh1 knockout mice (knockout restricted to neuronal expression from the beginning of development) | Impaired behavioral flexibility and extinction of previously consolidated memories, impaired LTP in hippocampal slices | [43] |
| Two loxP sites eliminate exons 2 and 3 from the Frz1 gene, cre recombinase expressed under the control of the forebrain-specific driver Emx | Conditional cdh1 knockout mice (knockout in neocortical and hippocampal excitatory neurons but not GABAergic interneurons) | Profoundly impaired induction of mGluR-dependent LTD in the hippocampus | [18] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchsberger, T.; Lloret, A.; Viña, J. New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1057. https://doi.org/10.3390/ijms18051057
Fuchsberger T, Lloret A, Viña J. New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease. International Journal of Molecular Sciences. 2017; 18(5):1057. https://doi.org/10.3390/ijms18051057
Chicago/Turabian StyleFuchsberger, Tanja, Ana Lloret, and Jose Viña. 2017. "New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease" International Journal of Molecular Sciences 18, no. 5: 1057. https://doi.org/10.3390/ijms18051057
APA StyleFuchsberger, T., Lloret, A., & Viña, J. (2017). New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease. International Journal of Molecular Sciences, 18(5), 1057. https://doi.org/10.3390/ijms18051057