
EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment



Abstract
:1. Introduction
1.1. Glioma Invasion and Angiogenesis
1.2. The Epidermal Growth Factor and the Mutant EGFRvIII
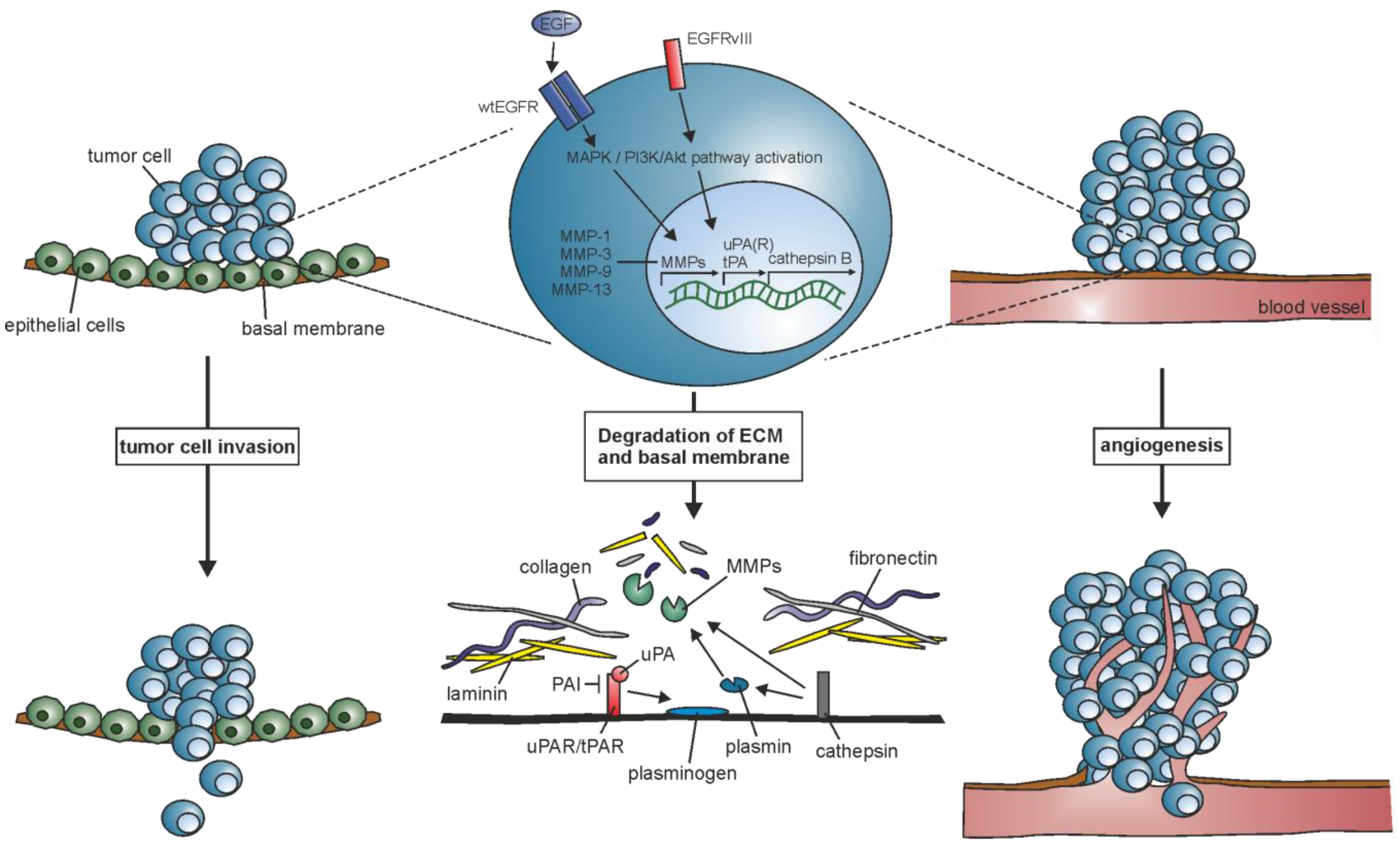
2. ECM Remodeling: Induction of Matrix-Degrading Proteases
2.1. Matrix Metalloprotease (MMPs)
2.1.1. EGFR and MMPs
2.1.2. EGFRvIII and MMPs
2.2. Serine Proteases
2.2.1. EGFR and Serine Proteases
2.2.2. EGFRvIII and Serine Proteases
2.3. Cysteine Proteases
2.3.1. EGFR and Cysteine Proteases
2.3.2. EGFRvIII and Cysteine Proteases
3. ECM-Cell Interaction: Activation of Transduction Signaling Pathways
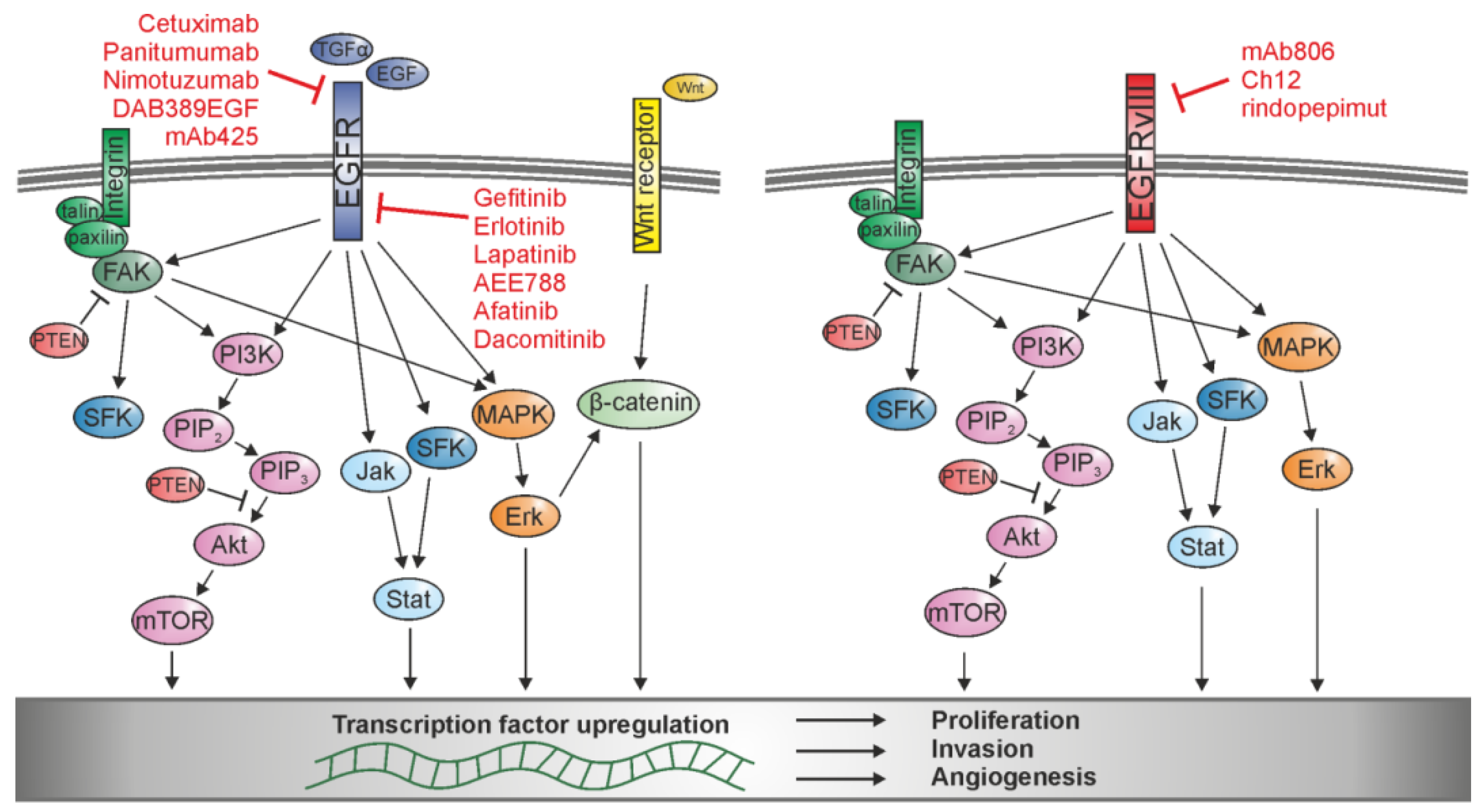
3.1. Integrin-FAK-ERK Signaling
3.1.1. EGFR in Integrin-FAK-ERK Signaling
3.1.2. EGFRvIII in Integrin-FAK-ERK Signaling
3.2. SFK Signaling
3.2.1. EGFR in SFK Signaling
3.2.2. EGFRvIII in SFK Signaling
3.3. JAK/STAT Signaling
3.3.1. EGFR in JAK/STAT Signaling
3.3.2. EGFRvIII in JAK/STAT Signaling
3.4. PI3K/Akt/mTOR Signaling
3.4.1. EGFR in PI3K/Akt/mTOR Signaling
3.4.2. EGFRvIII in PI3K/Akt/mTOR Signaling
3.5. Wnt/β-Catenin Signaling
3.5.1. EGFR in Wnt/β-Catenin Signaling
3.5.2. EGFRvIII in Wnt/β-Catenin Signaling
4. Combined Therapy as an Effective Approach for Glioma Treatment
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| EGFR | Epidermal growth factor receptor |
| GBM | Glioblastoma multiforme |
| ECM | Extracellular matrix |
| IDH | Isocitrate dehydrogenase |
| VEGF | Vascular endothelial growth factor receptor |
| EGF | Epidermal growth factor |
| FGF | Fibroblast growth factor |
| MMP | Matrix metalloprotease |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| GSC | Glioma stem-like cells |
| ERK | Extracellular-signal regulated kinase |
| uPA | Urokinase-type plasminogen activator |
| tPA | Tissue-type plasminogen activator |
| PAI | Plasminogen activator inhibitors |
| uPAR | Urokinase-type plasminogen activator receptor |
| PLCγ | Phospholipase Cγ |
| TGFα | Transforming growth factor |
| EGFL7 | EGF-like domain-containing protein 7 |
| Grb2 | Grb2 Growth factor receptor-bound protein 2 |
| PTEN | Phosphatase and tensin homolog |
| SFK | Src family kinase |
| JAK | Receptor-associated Janus kinase |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| mTOR | Mammalian target of rapamycin |
| GSK3 | Glycogen synthase kinase 3 |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Piantadosi, S.; Desideri, S.; Nabors, L.B.; Rosenfeld, M.; Fisher, J. Survival of Patients with Newly Diagnosed Glioblastoma Treated with Radiation and Temozolomide in Research Studies in the United States. Clin. Cancer Res. 2010, 16, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H. Structural development in gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.N.; Bigner, S.H. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat. Res. Rev. Genet. Toxicol. 1992, 276, 299–306. [Google Scholar] [CrossRef]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van Deurs, B.; Poulsen, H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Furnari, F.B.; Cavenee, W.K.; Bögler, O. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc. Natl. Acad. Sci. USA 2003, 100, 6505–6510. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Glazer, C.A.; Martinson, H.M.; Friedman, H.S.; Archer, G.E.; Sampson, J.H.; Riggins, G.J. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002, 62, 3335–3339. [Google Scholar] [PubMed]
- Penar, P.L.; Khoshyomn, S.; Bhushan, A.; Tritton, T.R. Inhibition of epidermal growth factor receptor-associated tyrosine kinase blocks glioblastoma invasion of the brain. Neurosurgery 1997, 40, 141–151. [Google Scholar] [PubMed]
- Guillamo, J.-S.; de Boüard, S.; Valable, S.; Marteau, L.; Leuraud, P.; Marie, Y.; Poupon, M.-F.; Parienti, J.-J.; Raymond, E.; Peschanski, M. Molecular mechanisms underlying effects of epidermal growth factor receptor inhibition on invasion, proliferation, and angiogenesis in experimental glioma. Clin. Cancer Res. 2009, 15, 3697–3704. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.; Vandenberg, S.; Cheng, S.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.; Cavenee, W.; Furnari, F. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Price, J.; Wilson, H.; Haites, N. Epidermal growth factor (EGF) increases the in vitro invasion, motility and adhesion interactions of the primary renal carcinoma cell line, A704. Eur. J. Cancer 1996, 32, 1977–1982. [Google Scholar] [CrossRef]
- Rosen, E.M.; Goldberg, I.D. Protein factors which regulate cell motility. Cell Dev. Biol. 1989, 25, 1079–1087. [Google Scholar] [CrossRef]
- Shibata, T.; Kawano, T.; Nagayasu, H.; Okumura, K.; Arisue, M.; Hamada, J.; Takeichi, N.; Hosokawa, M. Enhancing effects of epidermal growth factor on human squamous cell carcinoma motility and matrix degradation but not growth. Tumor Biol. 1996, 17, 168–175. [Google Scholar] [CrossRef]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Wikstrand, C.J.; Stanley, S.D.; Humphrey, P.A.; Pegram, C.N.; Archer, G.E.; Kurpad, S.; Shibuya, M.; Bigner, D.D. Investigation of a synthetic peptide as immunogen for a variant epidermal growth factor receptor associated with gliomas. J. Neuroimmunol. 1993, 46, 165–173. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—Impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Malkki, H. Trial Watch: Glioblastoma vaccine therapy disappointment in Phase III trial. Nat. Rev. Neurol. 2016, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.; Wong, H.; Laing, T.; Rewcastle, N.; Morris, D.; Muzik, H.; Leco, K.; Johnston, R.; Brasher, P.; Sutherland, G.; et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br. J. Cancer 1999, 79, 1828. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Steck, P.A.; Mohanam, S.; Stetler-Stevenson, W.G.; Liotta, L.A.; Sawaya, R. Elevated levels of Mr 92,000 type IV collagenase in human brain tumors. Cancer Res. 1993, 53, 2208–2211. [Google Scholar] [PubMed]
- Anand, M.; Van Meter, T.; Fillmore, H. Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J. Neuro Oncol. 2011, 104, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Maddirela, D.R.; Gujrati, M.; Rao, J. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 2012, 3, e445. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar] [PubMed]
- Choe, G.; Park, J.K.; Jouben-Steele, L.; Kremen, T.J.; Liau, L.M.; Vinters, H.V.; Cloughesy, T.F.; Mischel, P.S. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin. Cancer Res. 2002, 8, 2894–2901. [Google Scholar] [PubMed]
- Wang, F.; Xiao, W.; Sun, J.; Han, D.; Zhu, Y. MiRNA-181c inhibits EGFR-signaling-dependent MMP9 activation via suppressing Akt phosphorylation in glioblastoma. Tumor Biol. 2014, 35, 8653–8658. [Google Scholar] [CrossRef] [PubMed]
- Sangar, V.; Funk, C.C.; Kusebauch, U.; Campbell, D.S.; Moritz, R.L.; Price, N.D. Quantitative proteomic analysis reveals effects of epidermal growth factor receptor (EGFR) on invasion-promoting proteins secreted by glioblastoma cells. Mol. Cell. Proteom. 2014, 13, 2618–2631. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Jasti, S.L.; Kyritsis, A.P.; Yung, W.A.; Ali-Osman, F.; Nicolson, G.L.; Rao, J.S. Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by the extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clin. Exp. Metastasis 2000, 18, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lorimer, I.A.; Lavictoire, S.J. Activation of extracellular-regulated kinases by normal and mutant EGF receptors. Biochim. Biophy. Acta Mol. Cell Res. 2001, 1538, 1–9. [Google Scholar] [CrossRef]
- Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; Tan, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar]
- Jin, X.; Jin, X.; Sohn, Y.-W.; Yin, J.; Kim, S.-H.; Joshi, K.; Nam, D.-H.; Nakano, I.; Kim, H. Blockade of EGFR signaling promotes glioma stem-like cell invasiveness by abolishing ID3-mediated inhibition of p27 KIP1 and MMP3 expression. Cancer Lett. 2013, 328, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Rudnicki, A.; Smith, T.W. Expression of urokinase-type plasminogen activator receptor (uPAR) in primary central nervous system neoplasms. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Chandrasekar, N.; Kin, Y.; Lakka, S.S.; Mohanam, S.; Yanamandra, N.; Mohan, P.M.; Fuller, G.N.; Fang, B.; Fueyo, J.; et al. Suppression of glioma invasion and growth by adenovirus-mediated delivery of a bicistronic construct containing antisense uPAR and sense p16 gene sequences. Oncogene 2002, 21, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Czekay, R.-P.; Kuemmel, T.A.; Orlando, R.A.; Farquhar, M.G. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol. Biol. Cell 2001, 12, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Muracciole, X.; Romain, S.; Dufour, H.; Palmari, J.; Chinot, O.; Ouafik, L.H.; Grisoli, F.; Figarella, D.; Martin, P.-M. PAI-1 and EGFR expression in adult glioma tumors: Toward a molecular prognostic classification. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 592–598. [Google Scholar] [CrossRef]
- Kasza, A.; Kowanetz, M.; Poślednik, K.; Witek, B.; Kordula, T.; Koj, A. Epidermal growth factor and pro-inflammatory cytokines regulate the expression of components of plasminogen activation system in U373-MG astrocytoma cells. Cytokine 2001, 16, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Abe, T.; Wakabayashi, Y.; Hikawa, T.; Matsuo, K.-I.; Yamada, Y.; Kuwano, M.; Hori, S. Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-α or basic fibroblast growth factor. J. Neuro-Oncol. 2000, 46, 115–123. [Google Scholar] [CrossRef]
- Mamoune, A.; Kassis, J.; Kharait, S.; Kloeker, S.; Manos, E.; Jones, D.A.; Wells, A. DU145 human prostate carcinoma invasiveness is modulated by urokinase receptor (uPAR) downstream of epidermal growth factor receptor (EGFR) signaling. Exp. Cell Res. 2004, 299, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Paugh, S.W.; Bryan, L.; Kapitonov, D.; Wilczynska, K.M.; Gopalan, S.M.; Rokita, H.; Milstien, S.; Spiegel, S.; Kordula, T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCδ, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008, 22, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Jo, M.; Cavenee, W.K.; Furnari, F.; VandenBerg, S.R.; Gonias, S.L. Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acade. Sci. USA 2011, 108, 15984–15989. [Google Scholar] [CrossRef] [PubMed]
- Rempel, S.A.; Rosenblum, M.L.; Mikkelsen, T.; Yan, P.-S.; Ellis, K.D.; Golembieski, W.A.; Sameni, M.; Rozhin, J.; Ziegler, G.; Sloane, B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994, 54, 6027–6031. [Google Scholar] [PubMed]
- Gole, B.; Huszthy, P.C.; Popović, M.; Jeruc, J.; Ardebili, Y.S.; Bjerkvig, R.; Lah, T.T. The regulation of cysteine cathepsins and cystatins in human gliomas. Int. J. Cancer 2012, 131, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.; Alapati, K.; Malla, R.R.; Gondi, C.S.; Mohanam, S.; Dinh, D.H.; Rao, J.S. Mechanism of p27 upregulation induced by downregulation of cathepsin B and uPAR in glioma. Mol. Oncol. 2011, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal 2009, 2, re6. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, I.; Stanković, N.D.; Bicker, F.; Meister, J.; Braun, H.; Awwad, K.; Baumgart, J.; Simon, K.; Thal, S.C.; Patra, C.; et al. EGFL7 ligates αvβ3 integrin to enhance vessel formation. Blood 2013, 121, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Sato, T.; D’Avirro, N.; Morimoto, C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 1995, 182, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Appeddu, P.A.; Parsons, J.T.; Hildebrand, J.D.; Schaller, M.D.; Guan, J.-L. Interaction of focal adhesion kinase with cytoskeletal protein talin. J. Biol. Chem. 1995, 270, 16995–16999. [Google Scholar] [CrossRef] [PubMed]
- Rutka, J.T.; Muller, M.; Hubbard, S.L.; Forsdike, J.; Dirks, P.B.; Jung, S.; Tsugu, A.; Ivanchuk, S.; Costello, P.; Mondal, S.; et al. Astrocytoma adhesion to extracellular matrix: Functional significance of integrin and focal adhesion kinase expression. J. Neuropathol. Exp. Neurol. 1999, 58, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Guan, J.-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc.Natl. Acad. Sci. USA 1994, 91, 10148–10152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chattopadhyay, A.; Ji, Q.-S.; Owen, J.D.; Ruest, P.J.; Carpenter, G.; Hanks, S.K. Focal adhesion kinase promotes phospholipase C-γ1 activity. Proc.Natl. Acad. Sci. USA 1999, 96, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Teramoto, H.; Gutkind, J.S.; Yamada, K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: Roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996, 135, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Sieg, D.J.; Hauck, C.R.; Ilic, D.; Klingbeil, C.K.; Schaefer, E.; Damsky, C.H.; Schlaepfer, D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000, 2, 249–256. [Google Scholar] [PubMed]
- Lu, Z.; Jiang, G.; Blume-Jensen, P.; Hunter, T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol. 2001, 21, 4016–4031. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.B.; Moscatello, D.K.; Wong, A.J.; Cooper, J.A.; Stahl, W.L. Differential modulation of mitogen-activated protein (MAP) kinase/extracellular signal-related kinase kinase and MAP kinase activities by a mutant epidermal growth factor receptor. J. Biol. Chem. 1995, 270, 30562–30566. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 1999, 146, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.M.; Tao, B.B.; Wang, L.Y.; Liang, Y.L.; Jin, J.W.; Yang, Y.; Hu, Y.L.; Zha, X.L. Protein phosphatase activity of PTEN inhibited the invasion of glioma cells with epidermal growth factor receptor mutation type III expression. Int. J. Cancer 2005, 117, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Wang, C.; Sun, L.; Mei, C.; Yao, W.; Liu, Y.; Shi, Y.; Qiu, S.; Fan, J.; et al. The effect of epidermal growth factor receptor variant III on glioma cell migration by stimulating ERK phosphorylation through the focal adhesion kinase signaling pathway. Arch. Biochem. Biophys. 2010, 502, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Angers-Loustau, A.; Hering, R.; Werbowetski, T.E.; Kaplan, D.R.; Del Maestro, R.F. Src Regulates Actin Dynamics and Invasion of Malignant Glial Cells in Three Dimensions. Mol. Cancer Res. 2004, 2, 595–605. [Google Scholar] [PubMed]
- Huveldt, D.; Lewis-Tuffin, L.J.; Carlson, B.L.; Schroeder, M.A.; Rodriguez, F.; Giannini, C.; Galanis, E.; Sarkaria, J.N.; Anastasiadis, P.Z. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS ONE 2013, 8, e56505. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Thomas, S.M.; Xi, S.; Smithgall, T.E.; Siegfried, J.M.; Kamens, J.; Gooding, W.E.; Grandis, J.R. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004, 64, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK–Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, E.; Rosland, G.V.; Talasila, K.M.; Knappskog, S.; Keunen, O.; Sottoriva, A.; Foerster, S.; Solecki, G.; Taxt, T.; Jirik, R.; et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol. 2016, 18, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Zhu, S.; Cvrljevic, A.; Huang, T.T.; Sarkaria, S.; Ahkavan, D.; Dang, J.; Dinca, E.B.; Plaisier, S.B.; Oderberg, I.; et al. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009, 69, 6889–6898. [Google Scholar] [CrossRef] [PubMed]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [PubMed]
- Jarzynka, M.J.; Hu, B.; Hui, K.-M.; Bar-Joseph, I.; Gu, W.; Hirose, T.; Haney, L.B.; Ravichandran, K.S.; Nishikawa, R.; Cheng, S.-Y. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res. 2007, 67, 7203–7211. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Hu, B.; Jarzynka, M.J.; Li, Y.; Keezer, S.; Johns, T.G.; Tang, C.K.; Hamilton, R.L.; Vuori, K.; Nishikawa, R.; et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 3018–3023. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; Lucks, P.; Borghouts, C. The Function of Stat3 in Tumor Cells and Their Microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Liao, H.-J. Trafficking of receptor tyrosine kinases to the nucleus. Exp. Cell Res. 2009, 315, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A.; et al. TWIST is expressed in human gliomas, promotes invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Hsu, S.-C.; Xia, W.; Cao, X.; Shih, J.-Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.-C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Han, L.; Dong, Y.; Tian, J.; Huang, W.; Liu, Z.; Jia, X.; Jiang, T.; Zhang, J.; Li, X.; et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro Oncol. 2014, 16, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; DePinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Vara, J.Á.F.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.K.; Sharma, P.; Harbor, P.C.; Rahaman, S.O.; Haque, S.J. PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene 2005, 24, 7290–7300. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Tachibana, I.; Passe, S.M.; Huntley, B.K.; Borell, T.J.; Iturria, N.; O’fallon, J.R.; Schaefer, P.L.; Scheithauer, B.W.; James, C.D.; et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J. Natl. Cancer Inst. 2001, 93, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Viale, A.; Socci, N.D.; Wiedmann, M.; Hu, X.; Holland, E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 2003, 12, 889–901. [Google Scholar] [CrossRef]
- Purow, B.W.; Sundaresan, T.K.; Burdick, M.J.; Kefas, B.A.; Comeau, L.D.; Hawkinson, M.P.; Su, Q.; Kotliarov, Y.; Lee, J.; Zhang, W.; et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis 2008, 29, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Teodorczyk, M.; Schmidt, M.H. Notching on cancer’s door: Notch signaling in brain tumors. Front. Oncol. 2015, 4, 341. [Google Scholar] [CrossRef] [PubMed]
- Moscatello, D.K.; Holgado-Madruga, M.; Emlet, D.R.; Montgomery, R.B.; Wong, A.J. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yuan, M.; Kim, I.-A.; Chang, C.-M.; Bernhard, E.J.; Shu, H.-K.G. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 2004, 23, 4594–4602. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Nagane, M.; Mishima, K.; Huang, H.S.; Furnari, F.B.; Cavenee, W.K. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 2002, 62, 6764–6769. [Google Scholar] [PubMed]
- Choe, G.; Horvath, S.; Cloughesy, T.F.; Crosby, K.; Seligson, D.; Palotie, A.; Inge, L.; Smith, B.L.; Sawyers, C.L.; Mischel, P.S. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003, 63, 2742–2746. [Google Scholar] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Nagase, H.; Horii, A.; Miyoshi, Y.; Nakatsuru, S.; Aoki, T.; Arakawa, H.; Nakamura, Y.; Shimano, T.; Yanagisawa, A.; et al. Germ-line and somatic mutations of the APC gene in patients with turcot syndrome and analysis of APC mutations in brain tumors. Genes Chromosom. Cancer 1994, 9, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Dahmen, R.; Koch, A.; Denkhaus, D.; Tonn, J.; Sörensen, N.; Berthold, F.; Behrens, J.; Birchmeier, W.; Wiestler, O.; Pietsch, T. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001, 61, 7039–7043. [Google Scholar] [PubMed]
- Koch, A.; Hrychyk, A.; Hartmann, W.; Waha, A.; Mikeska, T.; Waha, A.; Schüller, U.; Sörensen, N.; Berthold, F.; Goodyer, C.G.; et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int. J. Cancer 2007, 121, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Pu, P.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H. Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009, 16, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Fang, D.; Litchfield, D.W.; et al. EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Mol. Cell 2009, 36, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lan, F.; Yang, W.; Yang, Y.; Han, L.; Zhang, A.; Liu, J.; Zeng, H.; Jiang, T.; Pu, P.; et al. Interruption of β-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010, 1366, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef]
- Han, L.; Yang, Y.; Yue, X.; Huang, K.; Liu, X.; Pu, P.; Jiang, H.; Yan, W.; Jiang, T.; Kang, C. Inactivation of PI3K/AKT signaling inhibits glioma cell growth through modulation of β-catenin-mediated transcription. Brain Res. 2010, 1366, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.S.; Eswaraiah, A.; Crombet, T.; Piedra, P.; Saurez, G.; Iyer, H.; Arvind, A. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. MAbs 2009, 1, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neuro Oncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Lakka, S.S.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004, 1, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681–4689. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014, 16, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Canadas, M.; Codony, J.; Hueto, J.; Arcas, A.; LLadó, A.; Puig, X.; Guix, M.; Raspall, G.; Albanell, J. Activated epidermal growth factor receptor: Studies in head and neck tumors and tumor cell lines after exposure to ligand and receptor tyrosine-kinase inhibitors. In Proceedings of the 35th Annual Meeting of the American Society of Clinical Oncology, Atlanta, GA, USA, 15–18 May 1999; p. 2392. [Google Scholar]
- Rhys-Evans, P.; Modjtahedi, H.; Court, W.; Box, G.; Eccles, S. Overexpression of epidermal growth factor receptor in human head and neck squamous carcinoma cell lines correlates with matrix metalloproteinase-9 expression and in vitro invasion. Int. J. Cancer 2000, 86, 307–317. [Google Scholar]
- Pornchai, O.; Rhys-Evans, P.; Box, G.M.; Eccles, S.A. Differential modulation of proliferation, matrix metalloproteinase expression and invasion of human head and neck squamous carcinoma cells by c-erbB ligands. Clin. Exp. Metastasis 1999, 17, 631–639. [Google Scholar]
- Matsumoto, T.; Perrotte, P.; Bar-Eli, M.; Inoue, K.; Kuniyasu, H.; Eve, B. Blockade of EGF-R signaling with anti-EGFR monoclonal antibody (Mab) C225 inhibits matrix metalloproteinase-9 (MMP-9) expression and invasion of human transitional cell carcinoma (TCC) in vitro and in vivo. Proc. Am. Assoc. Cancer Res. 1998, 39, 83. [Google Scholar]
- Goudar, R.K.; Shi, Q.; Hjelmeland, M.D.; Keir, S.T.; McLendon, R.E.; Wikstrand, C.J.; Reese, E.D.; Conrad, C.A.; Traxler, P.; Lane, H.A.; et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol. Cancer Ther. 2005, 4, 101–112. [Google Scholar] [PubMed]
- Xu, W.; Bi, Y.; Kong, J.; Zhang, J.; Wang, B.; Li, K.; Tian, M.; Pan, X.; Shi, B.; Gu, J.; et al. Combination of an anti-EGFRvIII antibody CH12 with Rapamycin synergistically inhibits the growth of EGFRvIII+PTEN− glioblastoma in vivo. Oncotarget 2016, 7, 24752. [Google Scholar] [PubMed]
- Talasila, K.M.; Soentgerath, A.; Euskirchen, P.; Rosland, G.V.; Wang, J.; Huszthy, P.C.; Prestegarden, L.; Skaftnesmo, K.O.; Sakariassen, P.Ø.; Eskilsson, E.; et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 2013, 125, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Therapy | Class | Targeting also | FDA Approval |
|---|---|---|---|---|
| EGFR 1 | Monoclonal antibodies | |||
| Cetuximab | Mouse-human chimeric antibody | HER1 | Colorectal cancer Squamous cell carcinoma of the head and neck | |
| Nimotuzumab | Human antibody | HER1 | Orphan status for glioma Squamous cell carcinoma of the head and neck | |
| Panitumumab | Human antibody | HER1 | Metastatic colorectal cancer | |
| 125 I-Mab 425 | Radiolabeled murine antibody | - | N/A 3 | |
| Immunotoxins | ||||
| DAB389EGF | EGFR-toxin fusion protein | - | N/A | |
| Small molecule inhibitors | ||||
| Gefitinib | Anilinoquinazoline-based reversible inhibitor | HER1 | Non-small cell lung cancer | |
| Erlotinib | Anilinoquinazoline-based reversible inhibitor | HER1 | Non-small cell lung cancer Pancreatic cancer | |
| Lapatinib | Thiazolylquinazoline-based reversible inhibitor | HER1/2 | HER2+ breast cancer | |
| Afatinib | Anilinoquinazoline-based reversible inhibitor | HER1/2/4 | Metastasized non-small cell lung cancer | |
| Dacomitinib | Anilinoquinazoline-based reversible inhibitor | HER1/2/4 | N/A | |
| AEE788 | Tyrosine kinase inhibitor | VEGFR 2, HER1/2, ErbB2 | N/A | |
| EGFRvIII | Monoclonal antibodies | |||
| mAb806 | Human antibody | - | N/A | |
| CH12 | Human antibody | - | N/A | |
| Vaccines | ||||
| Rindopepimut | Peptide vaccination | - | N/A |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. Int. J. Mol. Sci. 2017, 18, 1295. https://doi.org/10.3390/ijms18061295
Keller S, Schmidt MHH. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. International Journal of Molecular Sciences. 2017; 18(6):1295. https://doi.org/10.3390/ijms18061295
Chicago/Turabian StyleKeller, Stefanie, and Mirko H. H. Schmidt. 2017. "EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment" International Journal of Molecular Sciences 18, no. 6: 1295. https://doi.org/10.3390/ijms18061295
APA StyleKeller, S., & Schmidt, M. H. H. (2017). EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. International Journal of Molecular Sciences, 18(6), 1295. https://doi.org/10.3390/ijms18061295