Adipokines in Liver Cirrhosis



Abstract
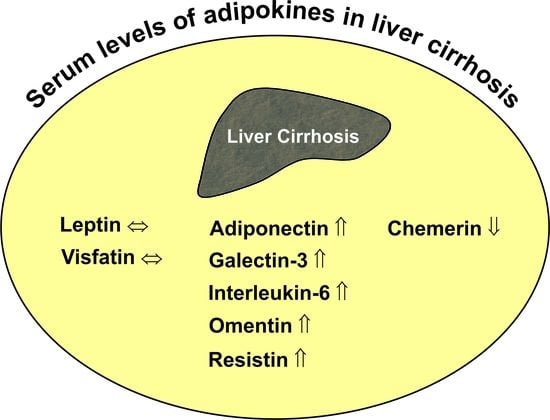
:
1. Introduction
2. Adipokines in Liver Cirrhosis
2.1. Adiponectin
2.1.1. General Information
2.1.2. Circulating Adiponectin in Liver Cirrhosis
2.2. Chemerin
2.2.1. General Information
2.2.2. Circulating Chemerin in Liver Cirrhosis
2.3. Leptin
2.3.1. General Information
2.3.2. Circulating Leptin in Liver Cirrhosis
2.4. Omentin
2.4.1. General Information
2.4.2. Circulating Omentin in Liver Cirrhosis
2.5. Galectin-3
2.5.1. General Information
2.5.2. Circulating Galectin-3 in Liver Cirrhosis
2.6. Resistin
2.6.1. General Information
2.6.2. Circulating Resistin in Liver Cirrhosis
2.7. Visfatin
2.7.1. General Information
2.7.2. Circulating Visfatin in Liver Cirrhosis
2.8. Interleukin-6
2.8.1. General Information
2.8.2. Circulating IL-6 in Liver Cirrhosis
3. Conclusions
4. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ge, P.S.; Runyon, B.A. Treatment of patients with cirrhosis. N. Engl. J. Med. 2016, 375, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol. 2011, 17, 2801–2811. [Google Scholar] [PubMed]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; Thiel, D.H.; Lewis, P.J.; Ramadori, G. Insulin production and resistance in different models of diet-induced obesity and metabolic syndrome. Int. J. Mol. Sci. 2017, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Marchesini, G.; Caracausi, L.; Macaluso, F.S.; Camma, C.; Ciminnisi, S.; Cabibi, D.; Porcasi, R.; Craxi, A.; di Marco, V. Industrial, not fruit fructose intake is associated with the severity of liver fibrosis in genotype 1 chronic hepatitis C patients. J. Hepatol. 2013, 59, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Berzigotti, A.; Garcia-Tsao, G.; Bosch, J.; Grace, N.D.; Burroughs, A.K.; Morillas, R.; Escorsell, A.; Garcia-Pagan, J.C.; Patch, D.; Matloff, D.S.; et al. Obesity is an independent risk factor for clinical decompensation in patients with cirrhosis. Hepatology 2011, 54, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Hickman, I.J.; Clouston, A.D.; Macdonald, G.A.; Purdie, D.M.; Prins, J.B.; Ash, S.; Jonsson, J.R.; Powell, E.E. Effect of weight reduction on liver histology and biochemistry in patients with chronic hepatitis C. Gut 2002, 51, 89–94. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.; Williams, R. Nutrition in end-stage liver disease: Principles and practice. Gastroenterology 2008, 134, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Alvares-da-Silva, M.R.; Reverbel da Silveira, T. Comparison between handgrip strength, subjective global assessment, and prognostic nutritional index in assessing malnutrition and predicting clinical outcome in cirrhotic outpatients. Nutrition 2005, 21, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T.; Shiraki, M.; Nishimura, K.; Ohnishi, S.; Imai, K.; Suetsugu, A.; Takai, K.; Shimizu, M.; Moriwaki, H. Sarcopenia impairs prognosis of patients with liver cirrhosis. Nutrition 2015, 31, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Tordjman, J.; Aron-Wisnewsky, J.; Poitou, C.; Oppert, J.M.; Torcivia, A.; Bouillot, J.L.; Paradis, V.; Ratziu, V.; Clement, K. Systematic review of bariatric surgery liver biopsies clarifies the natural history of liver disease in patients with severe obesity. Gut 2016, 24. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Schaffler, A. Does global gene expression analysis in type 2 diabetes provide an opportunity to identify highly promising drug targets? Endocr. Metab. Immune Disord. Drug Targets 2007, 7, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.T.; Pagliassotti, M.J. Metabolic alterations following visceral fat removal and expansion: Beyond anatomic location. Adipocyte 2012, 1, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Yates, K.P.; Vaughn, I.A.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; McCullough, A.; Merriman, R.; Hameed, B.; Doo, E.; Kleiner, D.E.; et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology 2013, 58, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.; Tsuchiya, K.; Tamaki, N.; Hirayama, I.; Tanaka, T.; Sato, M.; Yasui, Y.; Hosokawa, T.; Ueda, K.; Kuzuya, T.; et al. Effect of aging on risk for hepatocellular carcinoma in chronic hepatitis C virus infection. Hepatology 2010, 52, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Pares, A.; Caballeria, J.; Bruguera, M.; Torres, M.; Rodes, J. Histological course of alcoholic hepatitis. Influence of abstinence, sex and extent of hepatic damage. J. Hepatol. 1986, 2, 33–42. [Google Scholar] [CrossRef]
- Poynard, T.; Bedossa, P.; Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Peters, M.G. Liver disease in women: The influence of gender on epidemiology, natural history, and patient outcomes. Gastroenterol. Hepatol. 2013, 9, 633–639. [Google Scholar]
- D’Amico, G.; Garcia-Tsao, G.; Pagliaro, L. Natural history and prognostic indicators of survival in cirrhosis: A systematic review of 118 studies. J. Hepatol. 2006, 44, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Qi, X.; Guo, X. Child-pugh versus MELD score for the assessment of prognosis in liver cirrhosis: A systematic review and meta-analysis of observational studies. Medicine 2016, 95, e2877. [Google Scholar] [CrossRef] [PubMed]
- Pugh, R.N.; Murray-Lyon, I.M.; Dawson, J.L.; Pietroni, M.C.; Williams, R. Transection of the oesophagus for bleeding oesophageal varices. Br. J. Surg. 1973, 60, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Kamath, P.S.; Kim, W.R. The model for end-stage liver disease (MELD). Hepatology 2007, 45, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, M.; Moreau, R.; Angeli, P.; Schnabl, B.; Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 2015, 63, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Wiest, R. Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 353–372. [Google Scholar] [CrossRef] [PubMed]
- Van Thiel, D.H.; Alwahsh, S.M.; Ramadori, G. Metabolic Disease and Hepatocellular Carcinoma. In Hepatocellular Carcinoma: Diagnosis and Treatment; Carr, B.I., Ed.; Springer International Publishing: Cham, Vietnam, 2016; pp. 287–301. [Google Scholar]
- Ahmadieh, H.; Azar, S.T. Liver disease and diabetes: Association, pathophysiology, and management. Diabetes Res. Clin. Pract. 2014, 104, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Petrides, A.S.; Vogt, C.; Schulze-Berge, D.; Matthews, D.; Strohmeyer, G. Pathogenesis of glucose intolerance and diabetes mellitus in cirrhosis. Hepatology 1994, 19, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla-Bertot, L.; Vilar-Gomez, E.; Torres-Gonzalez, A.; Socias-Lopez, M.; Diago, M.; Adams, L.A.; Romero-Gomez, M. Impaired glucose metabolism increases risk of hepatic decompensation and death in patients with compensated hepatitis C virus-related cirrhosis. Dig. Liver Dis. 2016, 48, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Holland-Fischer, P.; Nielsen, M.F.; Vilstrup, H.; Tonner-Nielsen, D.; Mengel, A.; Schmitz, O.; Gronbaek, H. Insulin sensitivity and body composition in cirrhosis: Changes after TIPS. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G486–G493. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, J.; Alkhouri, N.; Dasarathy, S. Changes in body composition after transjugular intrahepatic portosystemic stent in cirrhosis: A critical review of literature. Liver Int. 2011, 31, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.F.; Tang, P.; Li, Q.; Yu, Z.T. Obesity, adipokines and hepatocellular carcinoma. Int. J. Cancer 2013, 133, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Stojsavljevic, S.; Gomercic Palcic, M.; Virovic Jukic, L.; Smircic Duvnjak, L.; Duvnjak, M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 18070–18091. [Google Scholar] [CrossRef] [PubMed]
- De Souza Batista, C.M.; Yang, R.Z.; Lee, M.J.; Glynn, N.M.; Yu, D.Z.; Pray, J.; Ndubuizu, K.; Patil, S.; Schwartz, A.; Kligman, M.; et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes 2007, 56, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Traber, P.G.; Zomer, E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 2013, 8, e83481. [Google Scholar] [CrossRef] [PubMed]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Streetz, K.L.; Tacke, F.; Leifeld, L.; Wustefeld, T.; Graw, A.; Klein, C.; Kamino, K.; Spengler, U.; Kreipe, H.; Kubicka, S.; et al. Interleukin 6/GP130-dependent pathways are protective during chronic liver diseases. Hepatology 2003, 38, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Ahima, R.S. Resistin in rodents and humans. Diabetes Metab. J. 2013, 37, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yang, X.; Liu, W.; Li, B.; Yin, W.; Shi, Y.; He, R. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene 2017, 36, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Su, L.; Esmaili, S.; Iseli, T.J.; Ramezani-Moghadam, M.; Hu, L.; Xu, A.; George, J.; Wang, J. Adiponectin attenuates liver fibrosis by inducing nitric oxide production of hepatic stellate cells. J. Mol. Med. 2015, 93, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Nepal, S.; Park, P.H. Modulation of cell death and survival by adipokines in the liver. Biol. Pharm. Bull. 2015, 38, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.; Wanninger, J.; Bauer, S.; Eisinger, K.; Neumeier, M.; Weiss, T.S.; Amann, T.; Hellerbrand, C.; Schaffler, A.; Scholmerich, J.; et al. Adiponectin reduces connective tissue growth factor in human hepatocytes which is already induced in non-fibrotic non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2011, 91, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Takehara, T.; Hayashi, N. Adipocytokines and liver disease. J. Gastroenterol. 2008, 43, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; Sanz-Garcia, C.; Nagy, L.E. Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr. Pathobiol. Rep. 2015, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H. The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Smith, T.; Rahman, K.; Thorn, N.E.; Anania, F.A. Adiponectin agonist ADP355 attenuates CCl4-induced liver fibrosis in mice. PLoS ONE 2014, 9, e110405. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin receptor binding proteins—Recent advances in elucidating adiponectin signalling pathways. FEBS Lett. 2010, 584, 4280–4286. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; Sifuentes, A.; Holland, W.L. Regulation of glucose and lipid homeostasis by adiponectin: Effects on hepatocytes, pancreatic β cells and adipocytes. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.C., 3rd; Brooks, J.S.; Lee, W.H.; Lee, M.G.; Kim, S.G. Therapeutic potential of dithiolethiones for hepatic diseases. Pharmacol. Ther. 2009, 124, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Traussnigg, S.A.; Trauner, M. Nuclear receptor modulation for the treatment of nonalcoholic fatty liver disease. Semin. Liver Dis. 2016, 36, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Halberg, N.; Schraw, T.D.; Wang, Z.V.; Kim, J.Y.; Yi, J.; Hamilton, M.P.; Luby-Phelps, K.; Scherer, P.E. Systemic fate of the adipocyte-derived factor adiponectin. Diabetes 2009, 58, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Balmer, M.L.; Joneli, J.; Schoepfer, A.; Stickel, F.; Thormann, W.; Dufour, J.F. Significance of serum adiponectin levels in patients with chronic liver disease. Clin. Sci. 2010, 119, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Kakizaki, S.; Sohara, N.; Yamazaki, Y.; Horiguchi, N.; Kanda, D.; Kabeya, K.; Katakai, K.; Sato, K.; Takagi, H.; Mori, M. Elevated plasma resistin concentrations in patients with liver cirrhosis. J. Gastroenterol. Hepatol. 2008, 23, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Kaser, S.; Moschen, A.; Kaser, A.; Ludwiczek, O.; Ebenbichler, C.F.; Vogel, W.; Jaschke, W.; Patsch, J.R.; Tilg, H. Circulating adiponectin reflects severity of liver disease but not insulin sensitivity in liver cirrhosis. J. Intern. Med. 2005, 258, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Kasztelan-Szczerbinska, B.; Surdacka, A.; Slomka, M.; Rolinski, J.; Celinski, K.; Smolen, A.; Szczerbinski, M. Association of serum adiponectin, leptin, and resistin concentrations with the severity of liver dysfunction and the disease complications in alcoholic liver disease. Mediat. Inflamm. 2013, 2013, 148526. [Google Scholar] [CrossRef] [PubMed]
- Salman, T.A.; Allam, N.; Azab, G.I.; Shaarawy, A.A.; Hassouna, M.M.; El-Haddad, O.M. Study of adiponectin in chronic liver disease and cholestasis. Hepatol. Int. 2010, 4, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Sohara, N.; Takagi, H.; Kakizaki, S.; Sato, K.; Mori, M. Elevated plasma adiponectin concentrations in patients with liver cirrhosis correlate with plasma insulin levels. Liver Int. 2005, 25, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Tietge, U.J.; Boker, K.H.; Manns, M.P.; Bahr, M.J. Elevated circulating adiponectin levels in liver cirrhosis are associated with reduced liver function and altered hepatic hemodynamics. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E82–E89. [Google Scholar] [CrossRef] [PubMed]
- Derbala, M.; Rizk, N.; Al-Kaabi, S.; Amer, A.; Shebl, F.; Al Marri, A.; Aigha, I.; Alyaesi, D.; Mohamed, H.; Aman, H.; et al. Adiponectin changes in HCV-Genotype 4: Relation to liver histology and response to treatment. J. Viral Hepat. 2009, 16, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Kalafateli, M.; Triantos, C.; Tsochatzis, E.; Michalaki, M.; Koutroumpakis, E.; Thomopoulos, K.; Kyriazopoulou, V.; Jelastopulu, E.; Burroughs, A.; Lambropoulou-Karatza, C.; et al. Adipokines levels are associated with the severity of liver disease in patients with alcoholic cirrhosis. World J. Gastroenterol. 2015, 21, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- Arano, T.; Nakagawa, H.; Tateishi, R.; Ikeda, H.; Uchino, K.; Enooku, K.; Goto, E.; Masuzaki, R.; Asaoka, Y.; Kondo, Y.; et al. Serum level of adiponectin and the risk of liver cancer development in chronic hepatitis C patients. Int. J. Cancer 2011, 129, 2226–2235. [Google Scholar] [CrossRef] [PubMed]
- Katira, A.; Tan, P.H. Evolving role of adiponectin in cancer-controversies and update. Cancer Biol. Med. 2016, 13, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Kitade, M.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Yamazaki, M.; et al. Crosstalk between high-molecular-weight adiponectin and T-cadherin during liver fibrosis development in rats. Int. J. Mol. Med. 2007, 20, 725–729. [Google Scholar] [PubMed]
- Abke, S.; Neumeier, M.; Weigert, J.; Wehrwein, G.; Eggenhofer, E.; Schaffler, A.; Maier, K.; Aslanidis, C.; Scholmerich, J.; Buechler, C. Adiponectin-induced secretion of interleukin-6 (IL-6), monocyte chemotactic protein-1 (MCP-1, CCL2) and interleukin-8 (IL-8, CXCL8) is impaired in monocytes from patients with type I diabetes. Cardiovasc. Diabetol. 2006, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G. Adiponectin in inflammatory and immune-mediated diseases. Cytokine 2013, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Weigert, J.; Obermeier, F.; Neumeier, M.; Wanninger, J.; Filarsky, M.; Bauer, S.; Aslanidis, C.; Rogler, G.; Ott, C.; Schaffler, A.; et al. Circulating levels of chemerin and adiponectin are higher in ulcerative colitis and chemerin is elevated in Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Wustefeld, T.; Horn, R.; Luedde, T.; Srinivas Rao, A.; Manns, M.P.; Trautwein, C.; Brabant, G. High adiponectin in chronic liver disease and cholestasis suggests biliary route of adiponectin excretion in vivo. J. Hepatol. 2005, 42, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Sadik, N.A.; Ahmed, A.; Ahmed, S. The significance of serum levels of adiponectin, leptin, and hyaluronic acid in hepatocellular carcinoma of cirrhotic and noncirrhotic patients. Hum. Exp. Toxicol. 2012, 31, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Moleda, L.; Farkas, S.; Scherer, M.; Kopp, A.; Wonckhaus, U.; Buchler, C.; Scholmerich, J.; Schaffler, A. Splanchnic concentrations and postprandial release of visceral adipokines. Metabolism 2010, 59, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Neumeier, M.; Hellerbrand, C.; Gabele, E.; Buettner, R.; Bollheimer, C.; Weigert, J.; Schaffler, A.; Weiss, T.S.; Lichtenauer, M.; Scholmerich, J.; et al. Adiponectin and its receptors in rodent models of fatty liver disease and liver cirrhosis. World J. Gastroenterol. 2006, 12, 5490–5494. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, K.L.; Sandahl, T.D.; Holland-Fischer, P.; Jessen, N.; Frystyk, J.; Flyvbjerg, A.; Gronbaek, H.; Vilstrup, H. Changes in adipokines after transjugular intrahepatic porto-systemic shunt indicate an anabolic shift in metabolism. Clin. Nutr. 2012, 31, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Mendes, F.; Lindor, K. Diagnostic model of esophageal varices in alcoholic liver disease. Eur. J. Gastroenterol. Hepatol. 2005, 17, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Ampuero, J.; Jover, M.; Abd-Elhalim, H.; Rincon, D.; Shatat, M.; Camacho, I.; Kamal, A.; Lo Iacono, O.; Nasr, Z.; et al. Predicting portal hypertension and variceal bleeding using non-invasive measurements of metabolic variables. Ann. Hepatol. 2013, 12, 588–598. [Google Scholar] [PubMed]
- Wiest, R.; Leidl, F.; Kopp, A.; Weigert, J.; Neumeier, M.; Buechler, C.; Schoelmerich, J.; Schaffler, A. Peritoneal fluid adipokines: Ready for prime time? Eur. J. Clin. Investig. 2009, 39, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.F.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Van der Poorten, D.; Samer, C.F.; Ramezani-Moghadam, M.; Coulter, S.; Kacevska, M.; Schrijnders, D.; Wu, L.E.; McLeod, D.; Bugianesi, E.; Komuta, M.; et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: Are alterations in serum adiponectin the cause? Hepatology 2013, 57, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Plauth, M.; Schutz, E.T. Cachexia in liver cirrhosis. Int. J. Cardiol. 2002, 85, 83–87. [Google Scholar] [CrossRef]
- Eisinger, K.; Krautbauer, S.; Wiest, R.; Weiss, T.S.; Buechler, C. Reduced serum chemerin in patients with more severe liver cirrhosis. Exp. Mol. Pathol. 2015, 98, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Eisinger, K.; Krautbauer, S.; Wiest, R.; Karrasch, T.; Hader, Y.; Scherer, M.N.; Farkas, S.; Aslanidis, C.; Buechler, C. Portal vein omentin is increased in patients with liver cirrhosis but is not associated with complications of portal hypertension. Eur. J. Clin. Investig. 2013, 43, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, J.; Weigert, J.; Wiest, R.; Bauer, S.; Karrasch, T.; Farkas, S.; Scherer, M.N.; Walter, R.; Weiss, T.S.; Hellerbrand, C.; et al. Systemic and hepatic vein galectin-3 are increased in patients with alcoholic liver cirrhosis and negatively correlate with liver function. Cytokine 2011, 55, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Weigert, J.; Wanninger, J.; Neumeier, M.; Bauer, S.; Schmidhofer, S.; Farkas, S.; Scherer, M.N.; Schaffler, A.; Scholmerich, J.; et al. Impaired hepatic removal of interleukin-6 in patients with liver cirrhosis. Cytokine 2011, 53, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.C.; Sinal, C.J. Chemerin: At the crossroads of inflammation and obesity. Trends Endocrinol. Metab. 2010, 21, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef] [PubMed]
- Krautbauer, S.; Wanninger, J.; Eisinger, K.; Hader, Y.; Beck, M.; Kopp, A.; Schmid, A.; Weiss, T.S.; Dorn, C.; Buechler, C. Chemerin is highly expressed in hepatocytes and is induced in non-alcoholic steatohepatitis liver. Exp. Mol. Pathol. 2013, 95, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Kwitniewski, M.; Banas, M.; Zabieglo, K.; Murzyn, K.; Cichy, J. Chemerin regulation and role in host defense. Am. J. Clin. Exp. Immunol. 2014, 3, 1–19. [Google Scholar] [PubMed]
- Buechler, C. Chemerin in Liver Diseases. Endocrinol. Metab. Syndr. 2014, 3, 1–6. [Google Scholar]
- Wanninger, J.; Bauer, S.; Eisinger, K.; Weiss, T.S.; Walter, R.; Hellerbrand, C.; Schaffler, A.; Higuchi, A.; Walsh, K.; Buechler, C. Adiponectin upregulates hepatocyte CMKLR1 which is reduced in human fatty liver. Mol. Cell. Endocrinol. 2012, 349, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Gruben, N.; Aparicio Vergara, M.; Kloosterhuis, N.J.; van der Molen, H.; Stoelwinder, S.; Youssef, S.; de Bruin, A.; Delsing, D.J.; Kuivenhoven, J.A.; van de Sluis, B.; et al. Chemokine-like receptor 1 deficiency does not affect the development of insulin resistance and nonalcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e96345. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Muruganandan, S.; Dranse, H.J.; McMullen, N.M.; Sinal, C.J. Gpr1 is an active chemerin receptor influencing glucose homeostasis in obese mice. J. Endocrinol. 2014, 222, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Okimura, Y.; Iguchi, G.; Nishizawa, H.; Yamamoto, M.; Suda, K.; Kitazawa, R.; Fujimoto, W.; Takahashi, K.; Zolotaryov, F.N.; et al. Chemerin regulates β-cell function in mice. Sci. Rep. 2011, 1, 123. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Rabe, K.; Lebherz, C.; Zugwurst, J.; Goke, B.; Parhofer, K.G.; Lehrke, M.; Broedl, U.C. Expression of human chemerin induces insulin resistance in the skeletal muscle but does not affect weight, lipid levels, and atherosclerosis in LDL receptor knockout mice on high-fat diet. Diabetes 2010, 59, 2898–2903. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Wanninger, J.; Schmidhofer, S.; Weigert, J.; Neumeier, M.; Dorn, C.; Hellerbrand, C.; Zimara, N.; Schaffler, A.; Aslanidis, C.; et al. Sterol regulatory element-binding protein 2 (SREBP2) activation after excess triglyceride storage induces chemerin in hypertrophic adipocytes. Endocrinology 2011, 152, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Weigert, J.; Neumeier, M.; Wanninger, J.; Filarsky, M.; Bauer, S.; Wiest, R.; Farkas, S.; Scherer, M.N.; Schaffler, A.; Aslanidis, C.; et al. Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin. Endocrinol. 2010, 72, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Toulany, J.; Parlee, S.D.; Sinal, C.J.; Slayter, K.; McNeil, S.; Goralski, K.B. CMKLR1 activation ex vivo does not increase proportionally to serum total chemerin in obese humans. Endocr. Connect. 2016, 5, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Eisenberg, D.; Zhao, L.; Adams, C.; Leib, R.; Morser, J.; Leung, L. Chemerin activation in human obesity. Obesity 2016, 24, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Kukla, M.; Zwirska-Korczala, K.; Gabriel, A.; Waluga, M.; Warakomska, I.; Szczygiel, B.; Berdowska, A.; Mazur, W.; Wozniak-Grygiel, E.; Kryczka, W. Chemerin, vaspin and insulin resistance in chronic hepatitis C. J. Viral Hepat. 2010, 17, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takai, K.; Hanai, T.; Shiraki, M.; Suzuki, Y.; Hayashi, H.; Naiki, T.; Nishigaki, Y.; Tomita, E.; Shimizu, M.; et al. Impact of serum chemerin levels on liver functional reserves and platelet counts in patients with hepatocellular carcinoma. Int. J. Mol. Sci. 2014, 15, 11294–11306. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A. Leptin signaling in the hypothalamus: Emphasis on energy homeostasis and leptin resistance. Front. Neuroendocrinol. 2003, 24, 225–253. [Google Scholar] [CrossRef] [PubMed]
- Catteau, A.; Caillon, H.; Barriere, P.; Denis, M.G.; Masson, D.; Freour, T. Leptin and its potential interest in assisted reproduction cycles. Hum. Reprod. Update 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; La Rocca, C.; Carbone, F.; de Rosa, V.; Galgani, M.; Matarese, G. Leptin as immune mediator: Interaction between neuroendocrine and immune system. Dev. Comp. Immunol. 2017, 66, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Aleffi, S.; Petrai, I.; Bertolani, C.; Parola, M.; Colombatto, S.; Novo, E.; Vizzutti, F.; Anania, F.A.; Milani, S.; Rombouts, K.; et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 2005, 42, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Ikejima, K.; Hirose, M.; Yoshikawa, M.; Lang, T.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology 2002, 36, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Farrell, G.C.; Schriemer, R.; Robertson, G.R. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [Google Scholar] [CrossRef]
- Schaffler, A.; Scholmerich, J.; Buchler, C. Mechanisms of disease: Adipocytokines and visceral adipose tissue—Emerging role in nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Bolukbas, F.F.; Bolukbas, C.; Horoz, M.; Gumus, M.; Erdogan, M.; Zeyrek, F.; Yayla, A.; Ovunc, O. Child-Pugh classification dependent alterations in serum leptin levels among cirrhotic patients: A case controlled study. BMC Gastroenterol. 2004, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J.; Bugianesi, E.; Marchesini, G.; Kalhan, S.C. Gender-dependent alterations in serum leptin in alcoholic cirrhosis. Gastroenterology 1998, 115, 947–953. [Google Scholar] [CrossRef]
- Ataseven, H.; Bahcecioglu, I.H.; Kuzu, N.; Yalniz, M.; Celebi, S.; Erensoy, A.; Ustundag, B. The levels of ghrelin, leptin, TNF-α, and IL-6 in liver cirrhosis and hepatocellular carcinoma due to HBV and HDV infection. Mediat. Inflamm. 2006, 2006, 78380. [Google Scholar] [CrossRef] [PubMed]
- Onodera, K.; Kato, A.; Suzuki, K. Serum leptin concentrations in liver cirrhosis: Relationship to the severity of liver dysfunction and their characteristic diurnal profiles. Hepatol. Res. 2001, 21, 205–212. [Google Scholar] [CrossRef]
- Shiraki, M.; Terakura, Y.; Iwasa, J.; Shimizu, M.; Miwa, Y.; Murakami, N.; Nagaki, M.; Moriwaki, H. Elevated serum tumor necrosis factor-α and soluble tumor necrosis factor receptors correlate with aberrant energy metabolism in liver cirrhosis. Nutrition 2010, 26, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Wang, Y.Y.; Sheu, W.H. Increased serum leptin concentrations correlate with soluble tumour necrosis factor receptor levels in patients with cirrhosis. Clin. Endocrinol. 2002, 57, 805–811. [Google Scholar] [CrossRef]
- Rachakonda, V.; Borhani, A.A.; Dunn, M.A.; Andrzejewski, M.; Martin, K.; Behari, J. Serum leptin is a biomarker of malnutrition in decompensated cirrhosis. PLoS ONE 2016, 11, e0159142. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Compean, D.; Jaquez-Quintana, J.O.; Lavalle-Gonzalez, F.J.; Gonzalez-Gonzalez, J.A.; Maldonado-Garza, H.J.; Villarreal-Perez, J.Z. Plasma cytokine levels imbalance in cirrhotic patients with impaired glucose tolerance and diabetes mellitus. A prospective study. Ann. Hepatol. 2014, 13, 403–410. [Google Scholar] [CrossRef]
- Campillo, B.; Sherman, E.; Richardet, J.P.; Bories, P.N. Serum leptin levels in alcoholic liver cirrhosis: Relationship with gender, nutritional status, liver function and energy metabolism. Eur. J. Clin. Nutr. 2001, 55, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Comlekci, A.; Akpinar, H.; Yesil, S.; Okan, I.; Ellidokuz, E.; Okan, A.; Ersoz, G.; Tankurt, E.; Batur, Y. Serum leptin levels in patients with liver cirrhosis and chronic viral hepatitis. Scand. J. Gastroenterol. 2003, 38, 779–786. [Google Scholar] [PubMed]
- Attar, B.M.; Moore, C.M.; George, M.; Ion-Nedelcu, N.; Turbay, R.; Zachariah, A.; Ramadori, G.; Fareed, J.; Van Thiel, D.H. Procalcitonin, and cytokines document a dynamic inflammatory state in non-infected cirrhotic patients with ascites. World J. Gastroenterol. 2014, 20, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Giannini, E.; Romagnoli, P.; Tenconi, G.L.; Botta, F.; Malfatti, F.; Chiarbonello, B.; Mamone, M.; Barreca, T.; Testa, R. High ascitic fluid leptin levels in patients with decompensated liver cirrhosis and sterile ascites: Relationship with TNF-α levels. Dig. Dis. Sci. 2004, 49, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Van Harmelen, V.; Reynisdottir, S.; Eriksson, P.; Thorne, A.; Hoffstedt, J.; Lonnqvist, F.; Arner, P. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes 1998, 47, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Nolte, W.; Wirtz, M.; Rossbach, C.; Leonhardt, U.; Buchwald, A.B.; Scholz, K.H.; Ramadori, G. TIPS implantation raises leptin levels in patients with liver cirrhosis. Exp. Clin. Endocrinol. Diabetes 2003, 111, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pray, J.; Wu, H.B.; Hansen, B.C.; Shuldiner, A.R.; Fried, S.K.; McLenithan, J.C.; Gong, D.W. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: Possible role in modulating insulin action. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1253–E1261. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, H.; Kuramoto, J.; Kameshima, S.; Usui, T.; Okada, M.; Hara, Y. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem. Biophys. Res. Commun. 2011, 408, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, S.; Shibata, R.; Kikuchi, R.; Izumiya, Y.; Rokutanda, T.; Araki, S.; Kataoka, Y.; Ohashi, K.; Daida, H.; Kihara, S.; et al. Fat-derived factor omentin stimulates endothelial cell function and ischemia-induced revascularization via endothelial nitric oxide synthase-dependent mechanism. J. Biol. Chem. 2012, 287, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, H.; Tsubaki, N.; Mukohda, M.; Okada, M.; Hara, Y. Omentin, a novel adipokine, induces vasodilation in rat isolated blood vessels. Biochem. Biophys. Res. Commun. 2010, 393, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R. Splanchnic and systemic vasodilation: The experimental models. J. Clin. Gastroenterol. 2007, 41 (Suppl. 3), S272–S287. [Google Scholar] [CrossRef] [PubMed]
- Laleman, W.; Landeghem, L.; Wilmer, A.; Fevery, J.; Nevens, F. Portal hypertension: From pathophysiology to clinical practice. Liver Int. 2005, 25, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Kukla, M.; Waluga, M.; Adamek, B.; Zalewska-Ziob, M.; Kasperczyk, J.; Gabriel, A.; Buldak, R.J.; Sobala-Szczygiel, B.; Kepa, L.; Ziora, K.; et al. Omentin serum concentration and hepatic expression in chronic hepatitis C patients-together or -apart? Pol. J. Pathol. 2015, 66, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Kukla, M.; Waluga, M.; Zorniak, M.; Berdowska, A.; Wosiewicz, P.; Sawczyn, T.; Buldak, R.J.; Ochman, M.; Ziora, K.; Krzeminski, T.; et al. Serum omentin and vaspin levels in cirrhotic patients with and without portal vein thrombosis. World J. Gastroenterol. 2017, 23, 2613–2624. [Google Scholar] [CrossRef] [PubMed]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Krautbauer, S.; Eisinger, K.; Hader, Y.; Buechler, C. Free fatty acids and IL-6 induce adipocyte galectin-3 which is increased in white and brown adipose tissues of obese mice. Cytokine 2014, 69, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Weigert, J.; Neumeier, M.; Wanninger, J.; Bauer, S.; Farkas, S.; Scherer, M.N.; Schnitzbauer, A.; Schaffler, A.; Aslanidis, C.; Scholmerich, J.; et al. Serum galectin-3 is elevated in obesity and negatively correlates with glycosylated hemoglobin in type 2 diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Valentim, I.B.; de Araujo, O.R.; Ataide Tda, R.; Goulart, M.O. Development of nonalcoholic hepatopathy: Contributions of oxidative stress and advanced glycation end products. Int. J. Mol. Sci. 2013, 14, 19846–19866. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Ricci, C.; Blasetti Fantauzzi, C.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.K.; Dowling, C.A.; Jeng, K.C.; Chen, J.T.; Yang, R.Y.; Liu, F.T. Galectin-3 expression is induced in cirrhotic liver and hepatocellular carcinoma. Int. J. Cancer 1999, 81, 519–526. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Liver fibrosis: From pathogenesis to novel therapies. Dig. Dis. 2016, 34, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Gudowska, M.; Gruszewska, E.; Cylwik, B.; Panasiuk, A.; Rogalska, M.; Flisiak, R.; Szmitkowski, M.; Chrostek, L. Galectin-3 concentration in liver diseases. Ann. Clin. Lab. Sci. 2015, 45, 669–673. [Google Scholar] [PubMed]
- Ulu, M.; Alacacioglu, A.; Yuksel, E.; Pamukk, B.O.; Bozkaya, G.; Ari, A.; Yuksel, A.; Sop, G.; Alacacioglu, I. Prognostic significance of serum galectin-3 levels in patients with hepatocellular cancer and chronic viral hepatitis. Saudi J. Gastroenterol. 2015, 21, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Obici, S.; Bhanot, S.; Monia, B.P.; McKay, R.A.; Rajala, M.W.; Scherer, P.E.; Rossetti, L. Role of resistin in diet-induced hepatic insulin resistance. J. Clin. Investig. 2004, 114, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.H.; Song, Y.; Ding, E.L.; Roberts, C.K.; Manson, J.E.; Rifai, N.; Buring, J.E.; Gaziano, J.M.; Liu, S. Circulating levels of resistin and risk of type 2 diabetes in men and women: Results from two prospective cohorts. Diabetes Care 2009, 32, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Eisinger, K.; Krautbauer, S. Diagnostic and prognostic potential of the macrophage specific receptor CD163 in inflammatory diseases. Inflamm. Allergy Drug Targets 2013, 12, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Bertolani, C.; Sancho-Bru, P.; Failli, P.; Bataller, R.; Aleffi, S.; DeFranco, R.; Mazzinghi, B.; Romagnani, P.; Milani, S.; Gines, P.; et al. Resistin as an intrahepatic cytokine: Overexpression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am. J. Pathol. 2006, 169, 2042–2053. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Zhao, C.Y.; Wang, W.; Wang, Y.D.; Sun, H.; Cao, W.; Yu, W.Y.; Zhang, L.; Ji, R.; Li, M.; et al. The relationship between hepatic resistin overexpression and inflammation in patients with nonalcoholic steatohepatitis. BMC Gastroenterol. 2014, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.X.; Su, L.; Brymora, J.; Bird, C.; Xie, Q.; George, J.; Wang, J.H. Resistin mediates the hepatic stellate cell phenotype. World J. Gastroenterol. 2013, 19, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Lin, C.Y.; Tsai, J.Y.; Wu, Y.L.; Su, K.H.; Lu, K.Y.; Hsiao, S.H.; Pan, C.C.; Kou, Y.R.; Hsu, Y.P.; et al. Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life Sci. 2009, 84, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.; Wilsie, L.; Palyha, O.; Strack, A.; Rashid, S. Discovery of a new role of human resistin in hepatocyte low-density lipoprotein receptor suppression mediated in part by proprotein convertase subtilisin/kexin type 9. J. Am. Coll. Cardiol. 2012, 59, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.H.; Di, J.; Jin, Y.; Zhang, Y.C.; Wu, M.; Sun, Y.; Zhang, G.Z. Resistin is expressed in human hepatocytes and induces insulin resistance. Endocrine 2008, 33, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bahr, M.J.; Ockenga, J.; Boker, K.H.; Manns, M.P.; Tietge, U.J. Elevated resistin levels in cirrhosis are associated with the proinflammatory state and altered hepatic glucose metabolism but not with insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E199–E206. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Zhang, Y.; Wei, Z.; Liu, P.; Kang, J.; Ma, D.; Ke, C.; Chen, Y.; Luo, J.; Gong, Z. High serum resistin associates with intrahepatic inflammation and necrosis: An index of disease severity for patients with chronic HBV infection. BMC Gastroenterol. 2017, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Yagmur, E.; Trautwein, C.; Gressner, A.M.; Tacke, F. Resistin serum levels are associated with insulin resistance, disease severity, clinical complications, and prognosis in patients with chronic liver diseases. Am. J. Gastroenterol. 2006, 101, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Dahl, T.B.; Haukeland, J.W.; Yndestad, A.; Ranheim, T.; Gladhaug, I.P.; Damas, J.K.; Haaland, T.; Loberg, E.M.; Arntsen, B.; Birkeland, K.; et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2010, 95, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Terra, X.; Auguet, T.; Quesada, I.; Aguilar, C.; Luna, A.M.; Hernandez, M.; Sabench, F.; Porras, J.A.; Martinez, S.; Lucas, A.; et al. Increased levels and adipose tissue expression of visfatin in morbidly obese women: The relationship with pro-inflammatory cytokines. Clin. Endocrinol. 2012, 77, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Berndt, J.; Kloting, N.; Kralisch, S.; Kovacs, P.; Fasshauer, M.; Schon, M.R.; Stumvoll, M.; Bluher, M. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes 2005, 54, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Petzold, S.; Barnikol-Oettler, A.; Korner, A.; Thasler, W.E.; Kratzsch, J.; Kiess, W.; Gebhardt, R. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem. Biophys. Res. Commun. 2010, 391, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Dahl, T.B.; Holm, S.; Aukrust, P.; Halvorsen, B. Visfatin/NAMPT: A multifaceted molecule with diverse roles in physiology and pathophysiology. Annu. Rev. Nutr. 2012, 32, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Choi, S.E.; Ha, E.S.; Kang, Y.; Han, S.J.; Kim, D.J.; Lee, K.W.; Kim, H.J. Extracellular visfatin activates gluconeogenesis in HepG2 cells through the classical PKA/CREB-dependent pathway. Horm. Metab. Res. 2014, 46, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Choi, S.E.; Ha, E.S.; Kang, Y.; Han, S.J.; Kim, D.J.; Lee, K.W.; Kim, H.J. Involvement of visfatin in palmitate-induced upregulation of inflammatory cytokines in hepatocytes. Metabolism 2011, 60, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.L.; Men, R.; Zhu, Y.; Yuan, C.; Wei, Y.; Liu, X.; Yang, L. Visfatin: An adipokine activator of rat hepatic stellate cells. Mol. Med. Rep. 2015, 11, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.M.; Kong, L.B.; Ren, W.G.; Wang, R.Q.; Du, J.H.; Li, W.C.; Zhao, S.X.; Zhang, Y.G.; Wu, W.J.; Di, H.L.; et al. Activation of peroxisome proliferator activated receptor α ameliorates ethanol mediated liver fibrosis in mice. Lipids Health Dis. 2013, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.F.; Bahr, M.J.; Boker, K.H.; Manns, M.P.; Tietge, U.J. Plasma levels of PBEF/Nampt/visfatin are decreased in patients with liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G196–G201. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhu, S.; Wu, Z.; Huang, Y.; Liu, C.; Tang, S.; Wei, L. Elevated serum visfatin levels are associated with poor prognosis of hepatocellular carcinoma. Oncotarget 2017, 8, 23427–23435. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.L. The relationship between serum visfatin and the progress of chronic viral hepatitis B cirrhosis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 297–301. [Google Scholar] [PubMed]
- Hammerich, L.; Tacke, F. Interleukins in chronic liver disease: Lessons learned from experimental mouse models. Clin. Exp. Gastroenterol. 2014, 7, 297–306. [Google Scholar] [PubMed]
- Klover, P.J.; Zimmers, T.A.; Koniaris, L.G.; Mooney, R.A. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 2003, 52, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, F.T.; Strohle, P.; Konner, A.C.; Gruber, S.; Tovar, S.; Bronneke, H.S.; Juntti-Berggren, L.; Li, L.S.; van Rooijen, N.; Libert, C.; et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010, 12, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Wustefeld, T.; Assmus, U.; Roskams, T.; Rose-John, S.; Muller, M.; Manns, M.P.; Ernst, M.; Trautwein, C. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J. Clin. Investig. 2005, 115, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Kovalovich, K.; DeAngelis, R.A.; Li, W.; Furth, E.E.; Ciliberto, G.; Taub, R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology 2000, 31, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Wuestefeld, T.; Klein, C.; Streetz, K.L.; Beraza, N.; Scholmerich, J.; Burgart, L.J.; Zender, L.; Kubicka, S.; Baskin-Bey, E.; Gores, G.J.; et al. Lack of GP130 expression results in more bacterial infection and higher mortality during chronic cholestasis in mice. Hepatology 2005, 42, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Andus, T.; Bauer, J.; Gerok, W. Effects of cytokines on the liver. Hepatology 1991, 13, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Goral, V.; Atalay, R.; Kucukoner, M. Insulin resistance in liver cirrhosis. Hepatogastroenterology 2010, 57, 309–315. [Google Scholar] [PubMed]
- Mortensen, C.; Andersen, O.; Krag, A.; Bendtsen, F.; Moller, S. High-sensitivity C-reactive protein levels predict survival and are related to haemodynamics in alcoholic cirrhosis. Eur. J. Gastroenterol. Hepatol. 2012, 24, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, A.; Gustot, T.; Durnez, A.; Evrard, S.; Moreno, C.; Quertinmont, E.; Vercruysse, V.; Demetter, P.; Franchimont, D.; Le Moine, O.; et al. An inhibitor of interleukin-6 trans-signalling, sgp130, contributes to impaired acute phase response in human chronic liver disease. Clin. Exp. Immunol. 2009, 156, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Zuwala-Jagiello, J.; Pazgan-Simon, M.; Simon, K.; Warwas, M. Advanced oxidation protein products and inflammatory markers in liver cirrhosis: A comparison between alcohol-related and HCV-related cirrhosis. Acta Biochim. Pol. 2011, 58, 59–65. [Google Scholar] [PubMed]
- Rao, R.K.; Seth, A.; Sheth, P. Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G881–G884. [Google Scholar] [CrossRef] [PubMed]
- Deviere, J.; Content, J.; Denys, C.; Vandenbussche, P.; Schandene, L.; Wybran, J.; Dupont, E. Excessive in vitro bacterial lipopolysaccharide-induced production of monokines in cirrhosis. Hepatology 1990, 11, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Agaugue, S.; Diaz, O.; Vanbervliet, B.; Dollet, S.; Guironnet-Paquet, A.; Andre, P.; Lotteau, V. Th1 disabled function in response to TLR4 stimulation of monocyte-derived DC from patients chronically-infected by hepatitis C virus. PLoS ONE 2008, 3, e2260. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Li, L.; Yang, E.N.; Dai, C.Y.; Liang, S.R.; Cao, W.K. Correlation between interleukin-6 and ammonia in patients with overt hepatic encephalopathy due to cirrhosis. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bachmann, R.A.; Chen, J. Interleukin-6 and insulin resistance. Vitam. Horm. 2009, 80, 613–633. [Google Scholar] [PubMed]
- Giron-Gonzalez, J.A.; Martinez-Sierra, C.; Rodriguez-Ramos, C.; Macias, M.A.; Rendon, P.; Diaz, F.; Fernandez-Gutierrez, C.; Martin-Herrera, L. Implication of inflammation-related cytokines in the natural history of liver cirrhosis. Liver Int. 2004, 24, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Dirchwolf, M.; Podhorzer, A.; Marino, M.; Shulman, C.; Cartier, M.; Zunino, M.; Paz, S.; Munoz, A.; Bocassi, A.; Gimenez, J.; et al. Immune dysfunction in cirrhosis: Distinct cytokines phenotypes according to cirrhosis severity. Cytokine 2016, 77, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes. Rev. 2013, 14, 245–262. [Google Scholar] [CrossRef] [PubMed]



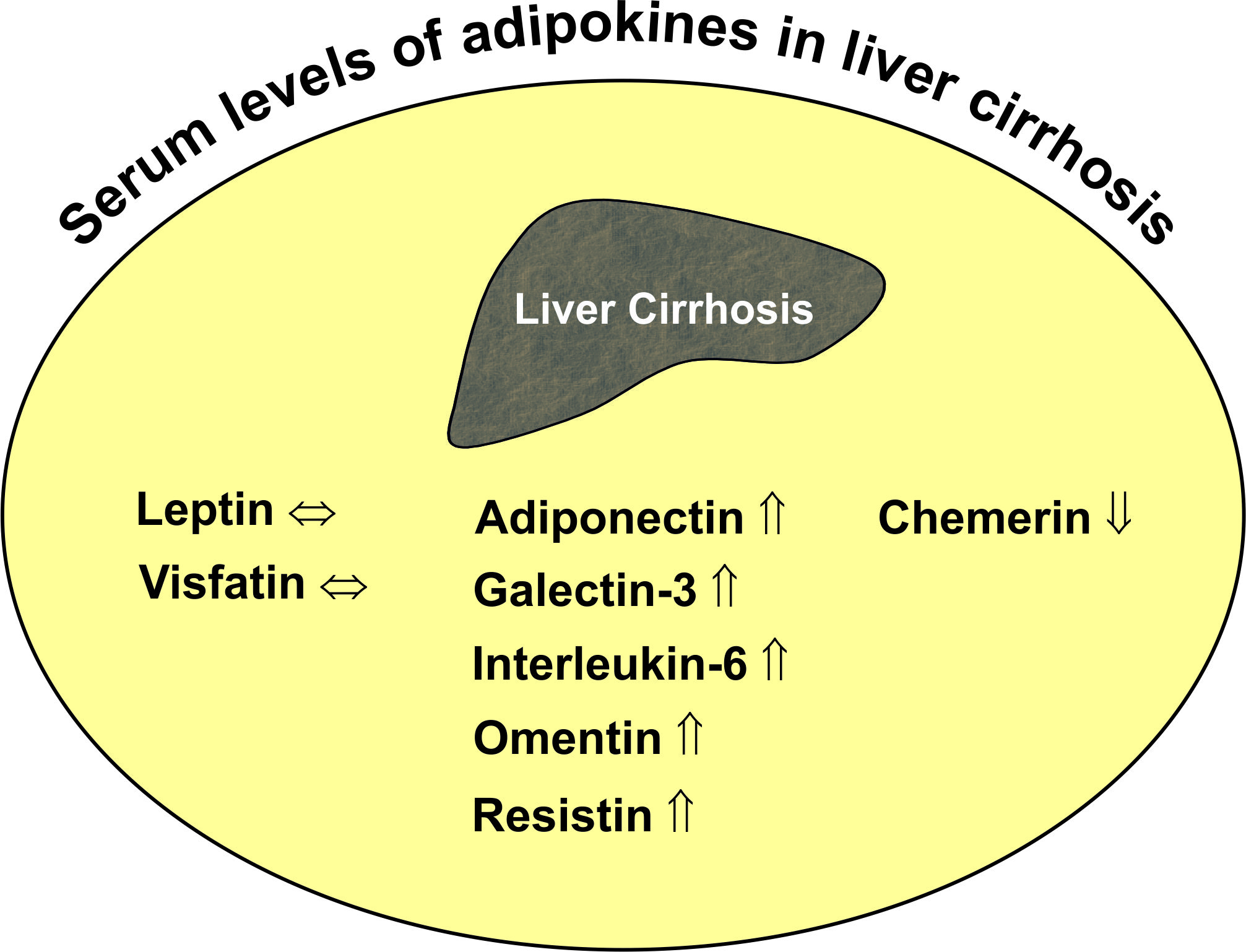{kind=link}
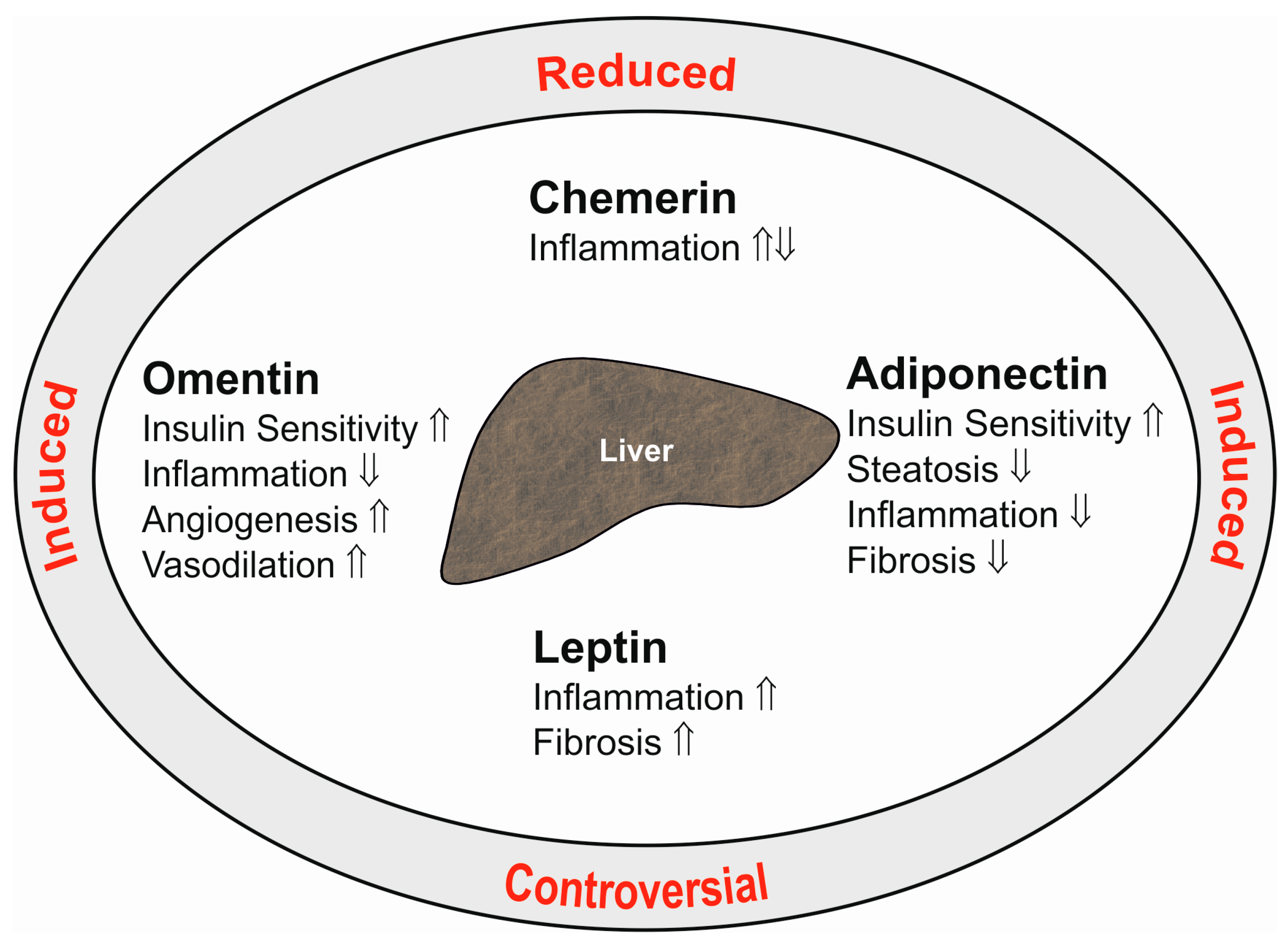{kind=link}
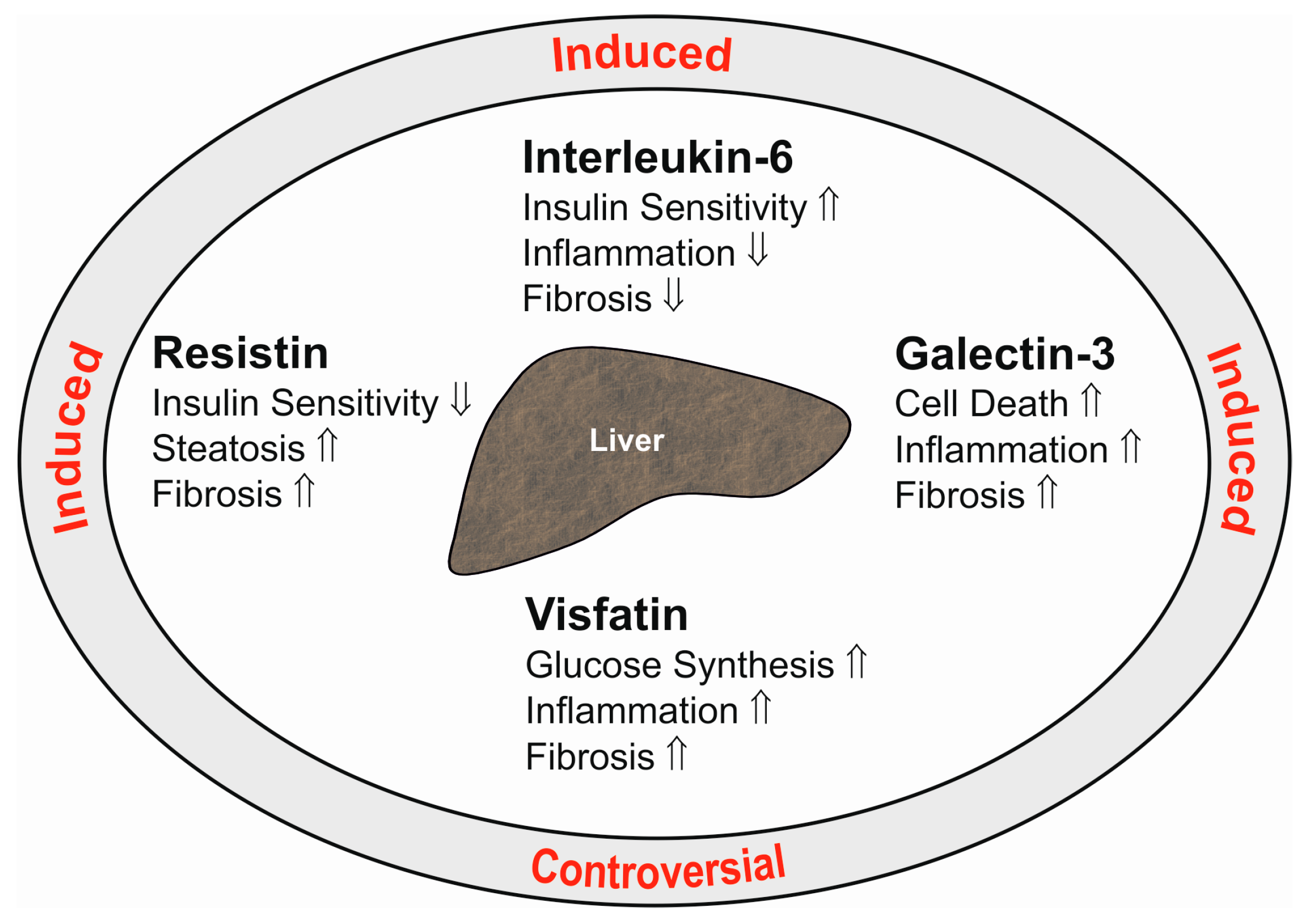{kind=link}
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Systemic Levels | Positive Associations with Scores | Reference |
|---|---|---|---|---|---|---|
| 147 (73) | 49.6 ± 11.9 | n.d. | Alcohol | ↑ HC | MELD, Child-Pugh | [60] |
| 45 (64) | 57 | 28.1 | Mixed | ↑ HC | n.d. | [57] |
| 38 (68) | 63 ± 15.5 | 21.9 ± 2.7 | Mixed | ↑ HC | Child-Pugh | [62] |
| 87 (51) | n.d. | n.d. | Mixed | ↑ HC | Child-Pugh | [59] |
| 40 (75) | 53.3 ± 8.2 | 27.9 ± 4.3 | HCV | ↑ HC | n.d. | [61] |
| 20 (75) | 47.6 ± 2.4 (SEM) | 23.0 ± 0.6 (SEM) | Mixed | ↑ HC | Child-Pugh | [63] |
| 79 (60) | 66.6 ± 8.9 | 22.3 ± 2.9 | HCV | ↑ HC | Child-Pugh | [58] |
| 40 (88) | 59 (37–75) | 25.89 (20.1–39.3) | Alcohol | n.d. | Child-Pugh | [65] |
| 93 (59) | n.d. | n.d. | Mixed | ↑ CLD | Highest in Child-Pugh B | [72] |
| 36 (64) | 53.0 ± 1.3 (SEM) | 27.1 ± 1.2 (SEM) | HCV | ↑ HC | n.d. | [73] |
| Adipokine | Cohort Size (% Male) | Age (Years) | BMI (kg/m2) | Etiology | SVS | HVS | PVS | References |
|---|---|---|---|---|---|---|---|---|
| Adiponectin | 11 (n.d.) | n.d. | n.d. | Viral | ≈ | ≈ | ≈ | [59] |
| Adiponectin | 50 (78) | 55 ± 2 | 25.5 ± 0.9 | Mostly alcoholic | ≈ | ≈ | ≈ | [74] |
| Chemerin | 45 (80) | 54 (26–81) | n.d. | Mostly alcoholic | ≈ | ↑ a | ≈ a | [83] |
| Leptin | 50 (78) | 55 ± 2 | 25.5 ± 0.9 | Mostly alcoholic | ↑ a | ≈ a | ≈ a | [74] |
| Omentin | 40 (80) | 52 (26–81) | 26.0 (16–38) | Mostly alcoholic | ≈ | ≈ a | ↑ a | [84] |
| Resistin | 50 (78) | 55 ± 2 | 25.5 ± 0.9 | Mostly alcoholic | ≈ | ≈ | ≈ | [74] |
| Galectin-3 | 33 (75.8) | 49 (40–81) | 25.6 (17–38) | Alcoholic | ≈ | ↑ a | ≈ a | [85] |
| Visfatin | 50 (78) | 55 ± 2 | 25.5 ± 0.9 | Mostly alcoholic | ≈ a,b | ↑ a | ↑ b | [74] |
| IL-6 | 41 (78) | n.d. | n.d. | Mostly alcoholic | ↑ a,b | ≈ a,b | ↑↑ a | [86] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Systemic Levels | Negative Associations with Scores | Reference |
|---|---|---|---|---|---|---|
| 44 (66) | 71 (50–82) | 22.5 (15.6–33.5) | Mostly HCV | n.d. | Child-Pugh | [103] |
| 45 (80) | 54 (26–81) | n.d. | Mostly alcohol | n.d. | Child-Pugh | [83] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Systemic Levels | Associations with Score | Reference |
|---|---|---|---|---|---|---|
| 107 (100) | 49.8 ± 11.5 | n.d. | Alcohol | Unchanged | No | [60] |
| 24 (66) | 64 (64–75) | 21.3 (17.3–26.9) | HCV | Unchanged | No | [115] |
| 13 (0) | 56.1 ± 7.4 | 22.6 ± 4.4 | Alcohol | Unchanged | No | [119] |
| 54 (56) | 55.5 (38.3–72.8) | 26.6 (20.6–32.7) | Mixed | n.d. | Child-Pugh negative | [118] |
| 36 (64) | 53.0 ± 1.3 (SEM) | 27.11 ± 1.18 (SEM) | HCV | Unchanged | n.d. | [73] |
| 40 (55) | 57 ± 11 | n.d. | Mixed | Unchanged | No | [114] |
| 18 (100) | 45 ± 1.5 (SEM) | 24.2 ± 0.8 (SEM) | Alcohol | Unchanged | No | [112] |
| 40 (88) | 59 (37–75) | 25.89 (20.1–39.3) | Alcohol | n.d. | No | [65] |
| 26 (100) | 59 (47–72) | 23.7 (21.9–25.5) | Mixed | ↑ | No | [116] |
| 10 (0) | 44 ± 3.7 (SEM) | 26.4 ± 2.2 (SEM) | Alcohol | ↑ | No | [112] |
| 35 (49) | 53 (28–73) | 24 (18–33) | HBV/HCV | ↑ | No | [111] |
| 24 (100) | 51.7 ± 10.6 | 22.3 ± 4.0 | Alcohol | ↑ | Child-Pugh positive | [119] |
| 24 (48) | 45.5 ± 8.0 | n.d. | HBV, HDV | ↓ | No | [113] |
| 40 (0) | 48.8 ± 9.9 | n.d. | Alcohol | ↓ | No | [60] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Systemic Levels | Associations with Scores | Reference |
|---|---|---|---|---|---|---|
| 40 (80.0) | 52 (26–81) | 26 (16–38) | Mostly alcohol | ↑ HC trend | No | [84] |
| 51 (68.6) | 52 (26–80) | 27 (17–40) | Mixed | n.d. | No | [132] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Serum Levels | Positive Associations with Scores | Reference |
|---|---|---|---|---|---|---|
| 87 (n.d.) | n.d. | n.d. | Mostly alcohol | ↑, HC Unchanged CLD | Child-Pugh | [141] |
| 22 (55) | 63.5 ± 10.8 | n.d. | HBV/HCV | ↑, CLD | n.d. | [142] |
| 33 (76) | 49 (40–81) | 25.6 (17–38) | Alcohol | ↑, HC | Child-Pugh MELD | [85] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Serum Levels | Positive Associations | Reference |
|---|---|---|---|---|---|---|
| 147 (72.8) | 49.6 ± 11.9 | n.d. | Alcohol | ↑ HC | No | [60] |
| 79 (59.5) | 66.6 ± 8.9 | 22.3 ± 2.9 | HCV | ↑ HC | Child-Pugh | [58] |
| 40 (87.5) | 59 (37–75) | 25.9 (20.1–39.3) | Alcohol | n.d. | No | [65] |
| 57 | 47.5 ± 1.3 (SEM) | 23.0 ± 0.4 (SEM) | Mixed | ↑ HC | Child-Pugh | [152] |
| 50 (78) | 55 ± 3 | 25.5 ± 0.9 | Mostly alcohol | ↑ HC | Child-Pugh | [74] |
| 70 (60) | 48.3 ± 11.1 | n.d. | HBV | ↑ HC, ↑ CLD | n.d. | [153] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Serum Levels | Associations | Reference |
|---|---|---|---|---|---|---|
| 19 (68) | 47.4 ± 2.5 (SEM) | 23.0 ± 0.6 (SEM) | Mixed | ↓ HC | No | [164] |
| 40 (87.5) | 59 (37–75) | 25.89 (20.08–39.31) | Alcoholic | n.d. | No | [65] |
| 129 (77.5) | 48.71 ± 11.65 | 22.86 ± 3.89 | HBV | ↑ HC, ↑ CLD | n.d. | [165] |
| 50 (78) | 55 ± 3 | 25.5 ± 0.9 | Mostly alcoholic | ↓ HC | Child-Pugh positive | [74] |
| 153 (59.5) | 42–60 | n.d. | HBV | ↑ HC | n.d. | [166] |
| Number of Patients (% Male) | Age (Years) | BMI (kg/m2) | Etiology | Serum Levels | Positive Associations in Patients | Reference |
|---|---|---|---|---|---|---|
| 41 (56) | 55 (40–81) | n.d. | Mixed | n.d. | Child-Pugh (trend) | [86] |
| 43 (63) | 56 (32–76) | n.d. | Alcohol | ↑ HC | Child-Pugh (trend) | [177] |
| 41 (63) | 58 (29–74) | 25.5 ± 0.9 | HCV | No change, HC | No | [177] |
| 70 (60) | 48.3 ± 11.1 | n.d. | HBV | ↑ HC, ↑ CLD | n.d. | [153] |
| 111 (60) | 46 (18–70) | n.d. | Mixed | ↑ HC, ↑ CLD | Child-Pugh | [40] |
| 79 (62) | 54.7 ± 14.7 | ↑ HC | n.d. | [174] | ||
| 72 (67) | 56.6 (36–75) | n.d. | HCV | ↑ HC | Child-Pugh (trend) | [39] |
| 72 (71) | 53 (46–60) | Mixed | No change, HC | Child-Pugh | [183] | |
| 45 (38) | n.d. | n.d. | Alcohol | ↑ (trend) HC | Child-Pugh (trend) | [175] |
| 14 (78.6) | 54 (44–59) | n.d. | Mostly HCV | ↑ HC | MELD | [184] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buechler, C.; Haberl, E.M.; Rein-Fischboeck, L.; Aslanidis, C. Adipokines in Liver Cirrhosis. Int. J. Mol. Sci. 2017, 18, 1392. https://doi.org/10.3390/ijms18071392
Buechler C, Haberl EM, Rein-Fischboeck L, Aslanidis C. Adipokines in Liver Cirrhosis. International Journal of Molecular Sciences. 2017; 18(7):1392. https://doi.org/10.3390/ijms18071392
Chicago/Turabian StyleBuechler, Christa, Elisabeth M. Haberl, Lisa Rein-Fischboeck, and Charalampos Aslanidis. 2017. "Adipokines in Liver Cirrhosis" International Journal of Molecular Sciences 18, no. 7: 1392. https://doi.org/10.3390/ijms18071392
APA StyleBuechler, C., Haberl, E. M., Rein-Fischboeck, L., & Aslanidis, C. (2017). Adipokines in Liver Cirrhosis. International Journal of Molecular Sciences, 18(7), 1392. https://doi.org/10.3390/ijms18071392