
Mitochondrial Dysfunction Mediated by Poly(ADP-Ribose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
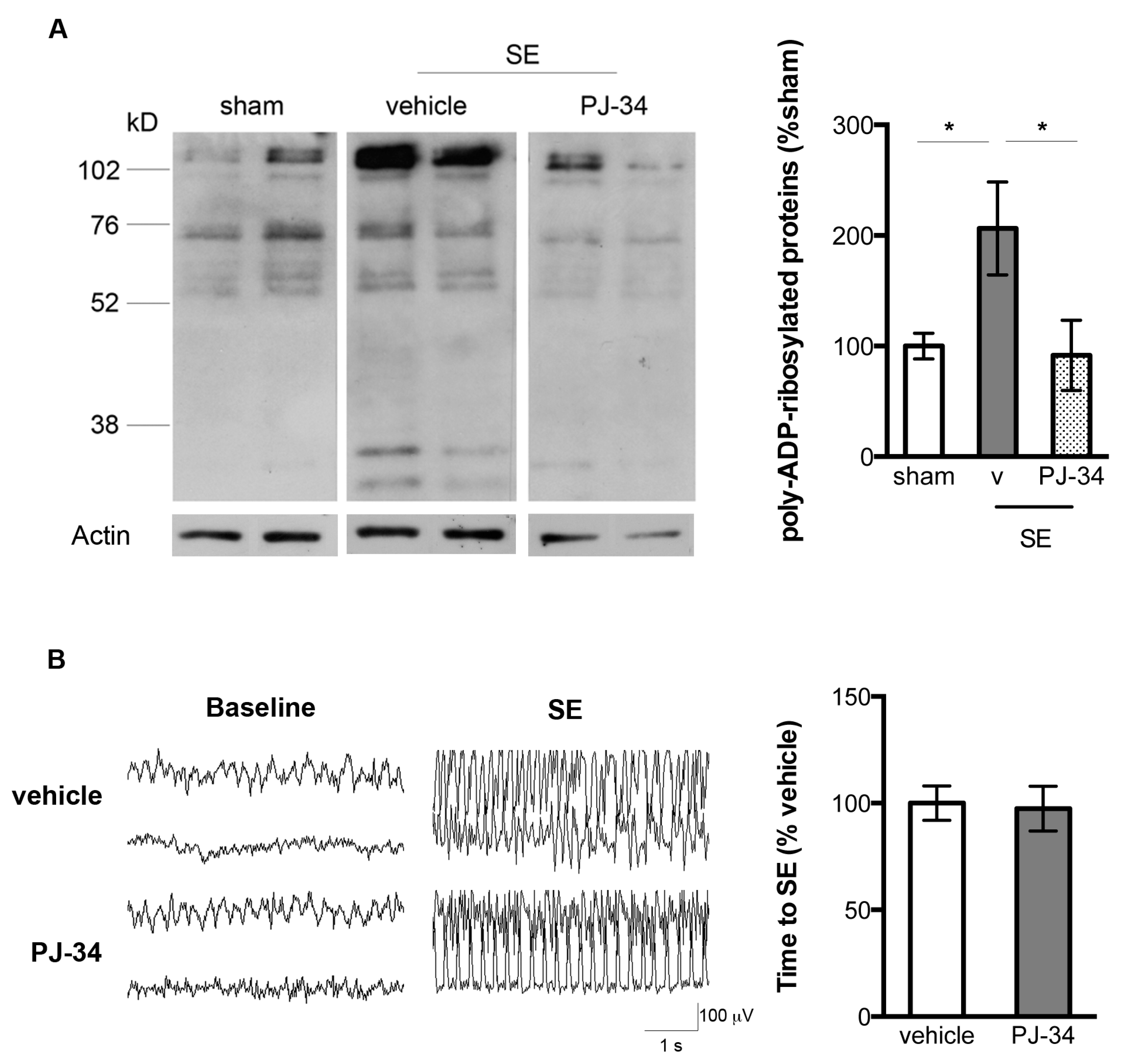
2.1. Increased PARP-1 Activity in the Hippocampus Following SE
2.2. PARP-1 Activation Following SE Was Associated with Intracellular NAD+ Depletion
2.3. PARP-1 Activation Following SE Was Associated Impaired NAD+-Dependent Mitochondrial Respiration
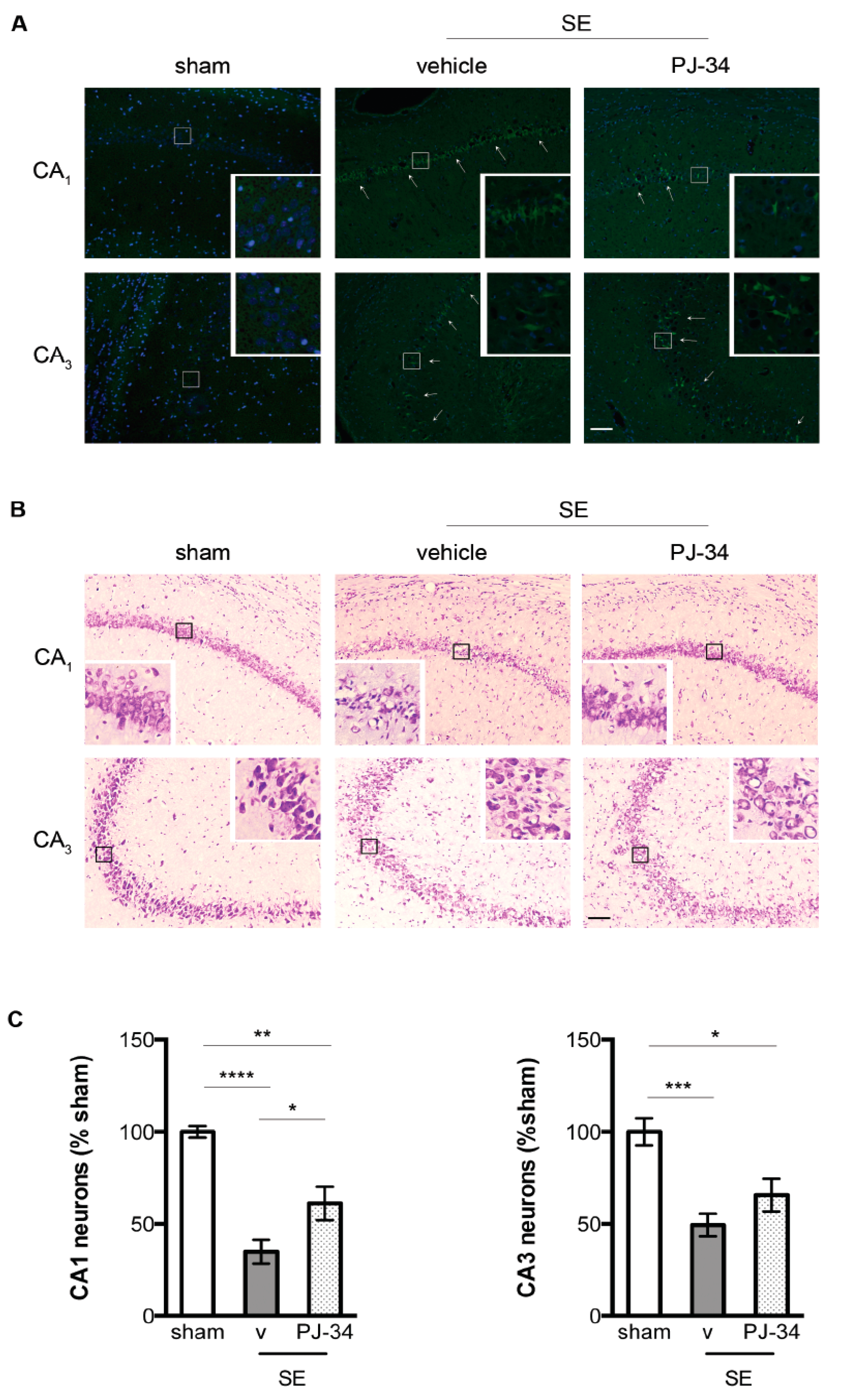
2.4. PARP-1 Activation Contributed to SE-Associated Hippocampal CA1 Neuronal Damage
3. Discussions
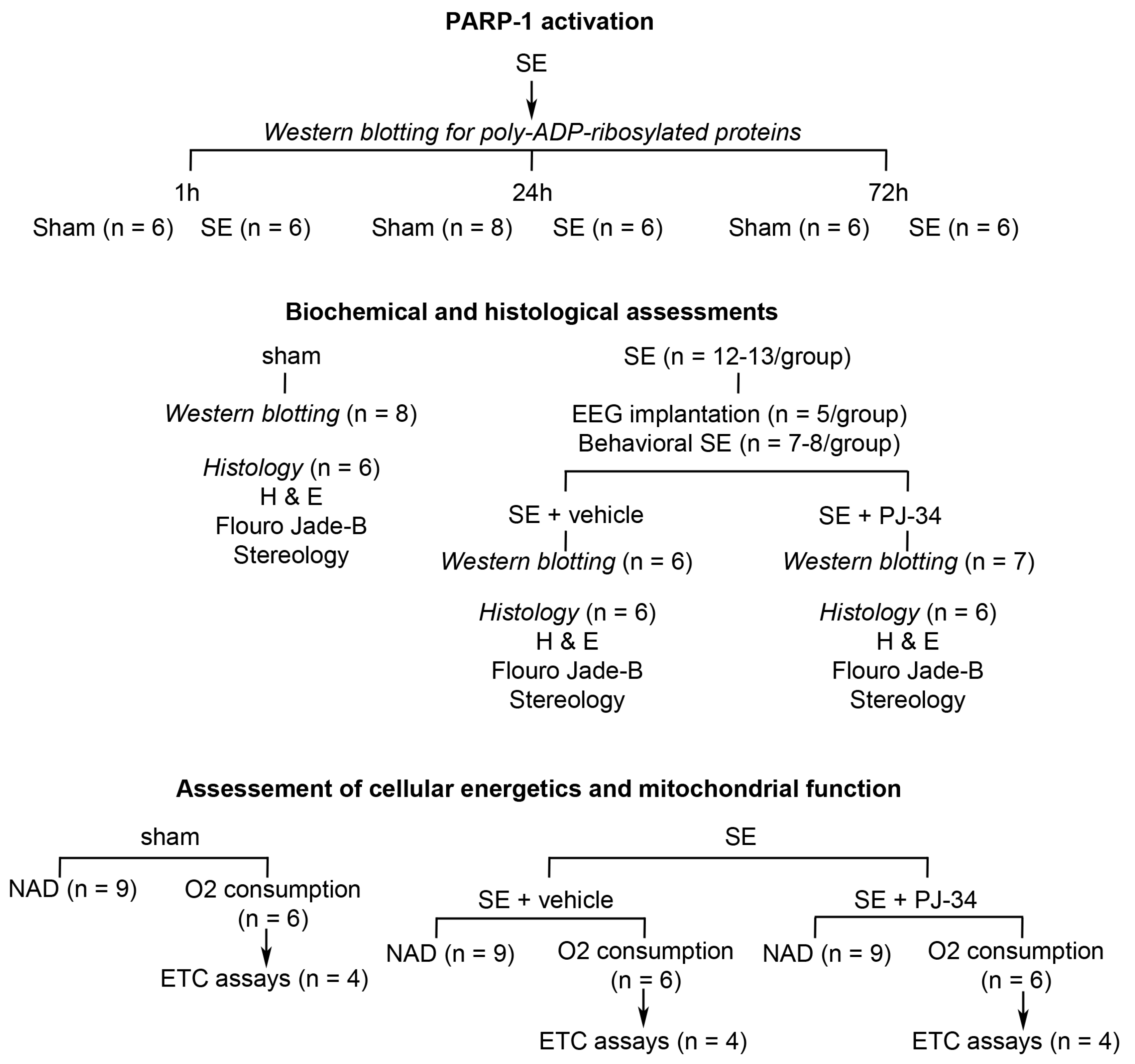
4. Materials and Methods
4.1. Kainic Acid-Induced SE
4.2. Stereotaxic EEG Implantation
4.3. pADPr Western Blotting
4.4. Mitochondrial Oxygen Consumption
4.5. Assessment of Electron Transport Chain Complex Activity
4.6. Tissue Preparation for Histological Studies
4.7. Fluoro-Jade B (FJ-B) Staining
4.8. Stereological Estimates of the Hippocampal Neurons
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticu—Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.V.; Shinnar, S.; Hesdorffer, D.C.; Bagiella, E.; Bello, J.A.; Chan, S.; Xu, Y.; MacFall, J.; Gomes, W.A.; Moshe, S.L.; et al. Hippocampal sclerosis after febrile status epilepticus: The FEBSTAT study. Ann. Neurol. 2014, 75, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, T.; Evans, M.C.; Meldrum, B.S. Intracellular calcium accumulation in rat hippocampus during seizures induced by bicuculline or L-allylglycine. Neuroscience 1983, 10, 385–395. [Google Scholar] [CrossRef]
- Williams, P.A.; White, A.M.; Clark, S.; Ferraro, D.J.; Swiercz, W.; Staley, K.J.; Dudek, F.E. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J. Neurosci. 2009, 29, 2103–2112. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Chang, A.Y.; Lin, J.W.; Hsu, S.P.; Chan, S.H. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia 2004, 45, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Kudina, T.A.; Seyfried, J.; Vielhaber, S.; Beck, H.; Elger, C.E.; Kunz, W.S. Seizure-dependent modulation of mitochondrial oxidative phosphorylation in rat hippocampus. Eur. J. Neurosci. 2002, 15, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef]
- Cock, H.R.; Tong, X.; Hargreaves, I.P.; Heales, S.J.; Clark, J.B.; Patsalos, P.N.; Thom, M.; Groves, M.; Schapira, A.H.; Shorvon, S.D.; et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002, 48, 157–168. [Google Scholar] [CrossRef]
- Rowley, S.; Liang, L.P.; Fulton, R.; Shimizu, T.; Day, B.; Patel, M. Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy. Neurobiol. Dis. 2015, 75, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Backos, D.S.; Reigan, P.; Patel, M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 2012, 32, 11250–11258. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blumcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Kovacs, R.; Schuchmann, S.; Gabriel, S.; Kardos, J.; Heinemann, U. Ca2+ signalling and changes of mitochondrial function during low-Mg2+-induced epileptiform activity in organotypic hippocampal slice cultures. Eur. J. Neurosci. 2001, 13, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Mandir, A.S.; Poitras, M.F.; Berliner, A.R.; Herring, W.J.; Guastella, D.B.; Feldman, A.; Poirier, G.G.; Wang, Z.Q.; Dawson, T.M.; Dawson, V.L. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J. Neurosci. 2000, 20, 8005–8011. [Google Scholar] [PubMed]
- Zhang, J.; Dawson, V.L.; Dawson, T.M.; Snyder, S.H. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science 1994, 263, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, S.W.; Koh, D.W.; Lew, J.; Coombs, C.; Bowers, W.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004, 24, 10963–10973. [Google Scholar] [CrossRef] [PubMed]
- Cosi, C.; Cavalieri, E.; Carcereri de Prati, A.; Marien, M.; Suzuki, H. Effects of kainic acid lesioning on poly(ADP-ribose) polymerase (PARP) activity in the rat striatum in vivo. Amino Acids 2000, 19, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Cosi, C.; Guerin, K.; Marien, M.; Koek, W.; Rollet, K. The PARP inhibitor benzamide protects against kainate and NMDA but not AMPA lesioning of the mouse striatum in vivo. Brain Res. 2004, 996, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Wang, S.H.; Song, Z.F.; Liu, X.W.; Wang, R.; Chi, Z.F. Poly(ADP-ribose) polymerase inhibitor is neuroprotective in epileptic rat via apoptosis-inducing factor and Akt signaling. Neuroreport 2007, 18, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, S.; Lin, Y.; Han, Y.; Qiu, X.; Zhao, X.; Cao, L.; Wang, X.; Chi, Z. Poly(ADP-ribose) polymerase inhibition protects epileptic hippocampal neurons from apoptosis via suppressing Akt-mediated apoptosis-inducing factor translocation in vitro. Neuroscience 2013, 231, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Hayaishi, O. ADP-ribosylation. Annu. Rev. Biochem. 1985, 54, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004, 279, 18895–18902. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [PubMed]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Chen, Y.; Watkins, S.C.; Nathaniel, P.D.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Szabo, C.; Clark, R.S. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 2007, 104, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Cosi, C.; Marien, M. Decreases in mouse brain NAD+ and ATP induced by 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP): Prevention by the poly(ADP-ribose) polymerase inhibitor, benzamide. Brain Res. 1998, 809, 58–67. [Google Scholar] [CrossRef]
- Hagberg, H.; Wilson, M.A.; Matsushita, H.; Zhu, C.; Lange, M.; Gustavsson, M.; Poitras, M.F.; Dawson, T.M.; Dawson, V.L.; Northington, F.; et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J. Neurochem. 2004, 90, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985, 14, 375–403. [Google Scholar] [CrossRef]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 1955, 217, 383–393. [Google Scholar] [PubMed]
- Krietsch, J.; Rouleau, M.; Pic, E.; Ethier, C.; Dawson, T.M.; Dawson, V.L.; Masson, J.Y.; Poirier, G.G.; Gagne, J.P. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Asp. Med. 2013, 34, 1066–1087. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Lubin, F.D.; Johnston, L.D.; Sweatt, J.D.; Anderson, A.E. Kainate mediates nuclear factor-kappa B activation in hippocampus via phosphatidylinositol-3 kinase and extracellular signal-regulated protein kinase. Neuroscience 2005, 133, 969–981. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Slomianka, L.; Gundersen, H.J. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991, 231, 482–497. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, Y.-C.; Baker, J.S.; Donti, T.; Graham, B.H.; Craigen, W.J.; Anderson, A.E. Mitochondrial Dysfunction Mediated by Poly(ADP-Ribose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus. Int. J. Mol. Sci. 2017, 18, 1502. https://doi.org/10.3390/ijms18071502
Lai Y-C, Baker JS, Donti T, Graham BH, Craigen WJ, Anderson AE. Mitochondrial Dysfunction Mediated by Poly(ADP-Ribose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus. International Journal of Molecular Sciences. 2017; 18(7):1502. https://doi.org/10.3390/ijms18071502
Chicago/Turabian StyleLai, Yi-Chen, J. Scott Baker, Taraka Donti, Brett H. Graham, William J. Craigen, and Anne E. Anderson. 2017. "Mitochondrial Dysfunction Mediated by Poly(ADP-Ribose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus" International Journal of Molecular Sciences 18, no. 7: 1502. https://doi.org/10.3390/ijms18071502
APA StyleLai, Y. -C., Baker, J. S., Donti, T., Graham, B. H., Craigen, W. J., & Anderson, A. E. (2017). Mitochondrial Dysfunction Mediated by Poly(ADP-Ribose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus. International Journal of Molecular Sciences, 18(7), 1502. https://doi.org/10.3390/ijms18071502