Finding a Balance between Protection and Pathology: The Dual Role of Perforin in Human Disease

Abstract
:1. Introduction
2. Perforin Evolution, Structure, and Function
2.1. Perforin Evolution
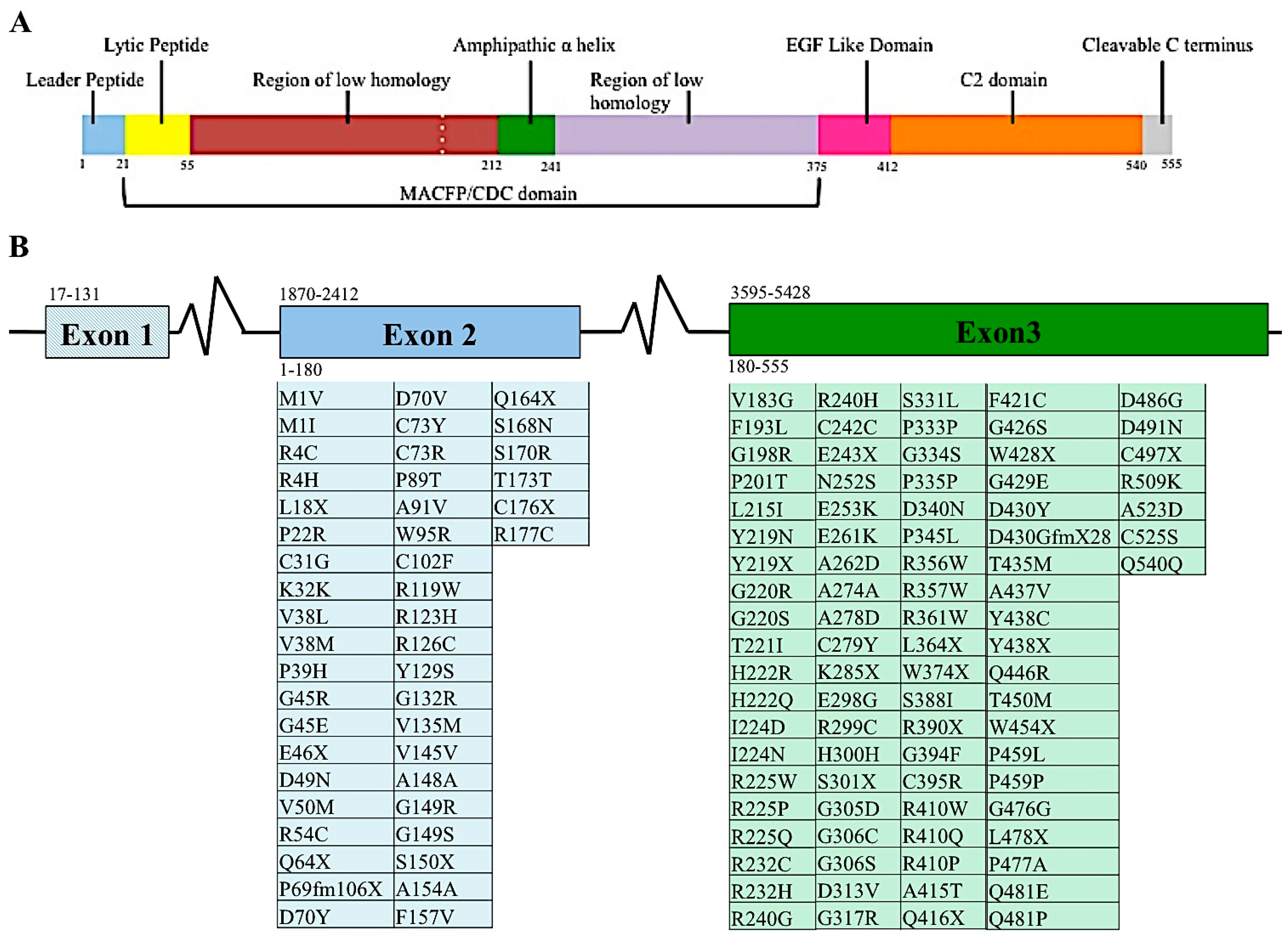
2.2. Genetic and Proteome Organization of Perforin
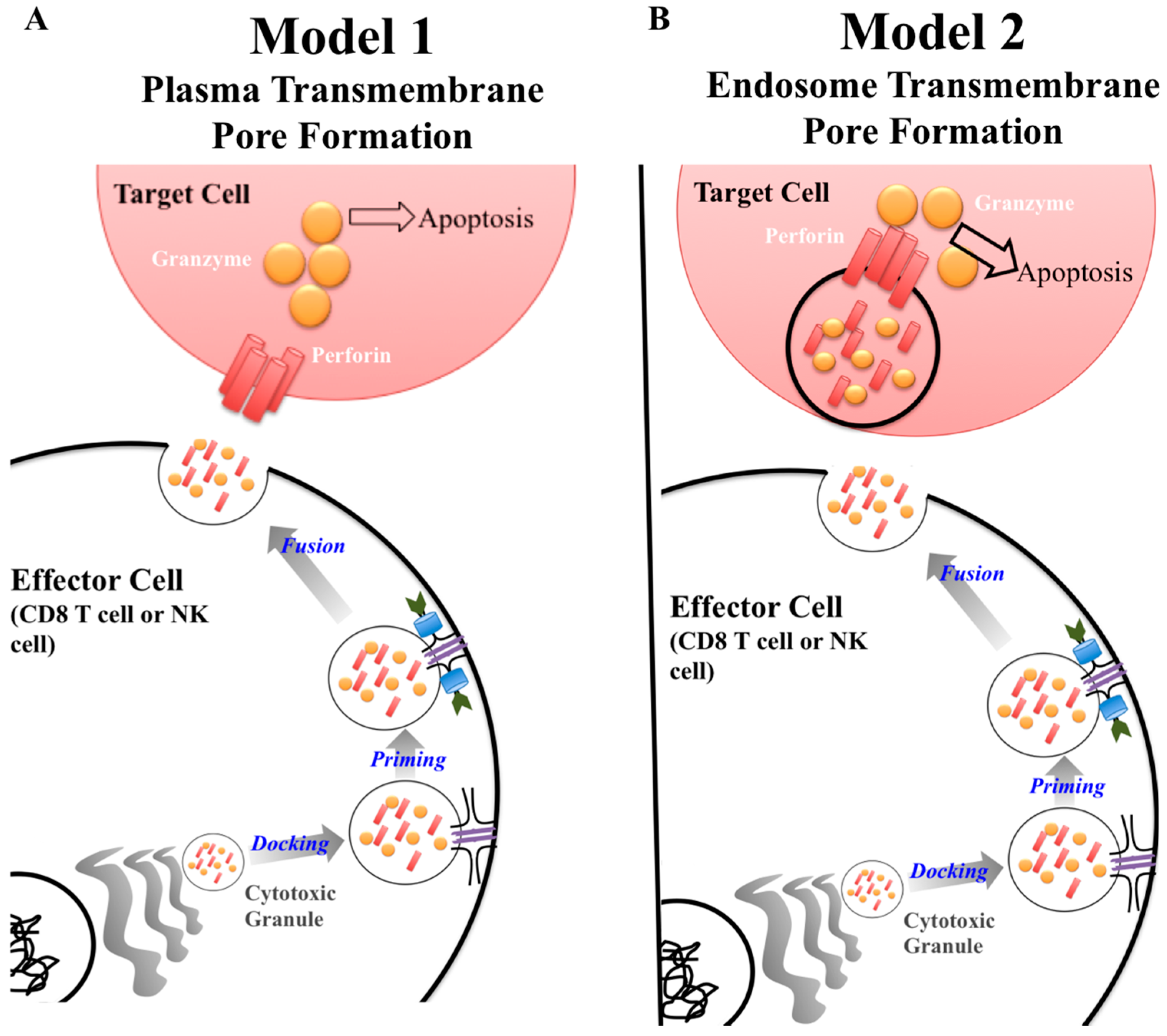
2.3. Discovery of Perforin as a Key Component of Cytolytic Killing
2.4. Perforin Single Nucleotide Variants (SNVs)
3. Human Disease Relevance of Perforin Single Nucleotide Variants
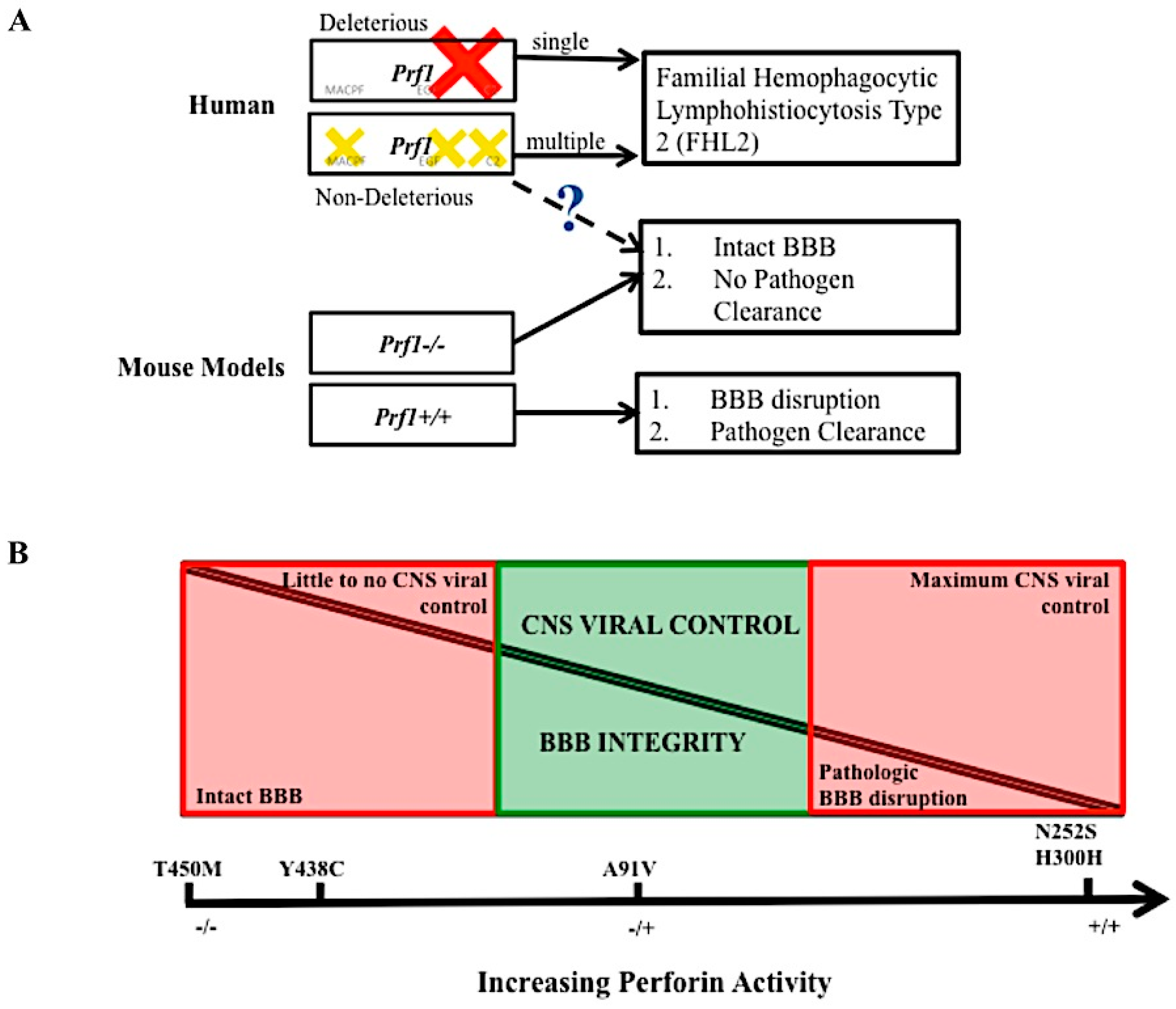
3.1. Primary and Secondary Hemophagocytic Lymphohistiocytosis
3.2. FHL Prevalence
3.3. FHL 2 Incidence
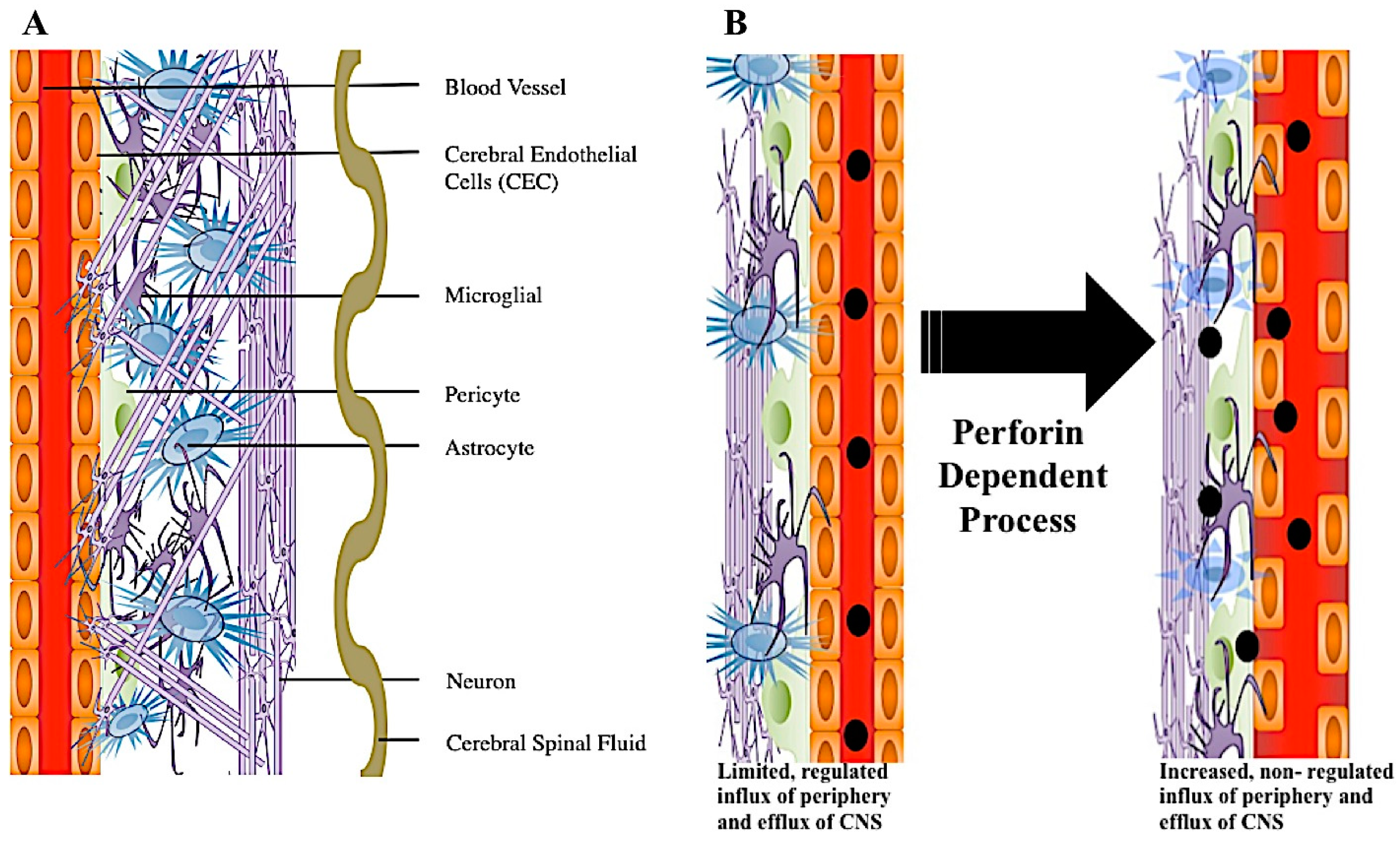
4. A Role for Perforin in Blood–Brain Barrier Disruption
The Integrity and Disruption of the Blood–Brain Barrier
5. Conclusions and Remaining Questions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FHL 2 | Familial hemophagocytic lymphohistiocytosis type 2 |
| BBB | Blood–brain barrier |
| SNV | Single nucleotide variant |
| NK | Natural Killer |
| CTL | CD8+ cytotoxic T lymphocytes |
| MHC I | Major histocompatibility complex class I |
| KIR | Killer cell immunoglobulin receptor |
| KLR | Killer cell lectin like receptor |
| CNS | Central nervous system |
| VEGF | Vascular endothelial growth factor |
| MPEG1 | Macrophage expressed gene/protein 1 |
| MACPF/CDC | Membrane attach complex perforin like/cholesteral dependent cytosylin |
| MAC | Membrane attack complex |
| EGF | Epidermal growth factor |
| MS | Multiple sclerosis |
| ALPS | Autoimmune lymphoproliferative syndrome |
| HLH | Hemophagocytic Lymphohistiocytosis |
| EBV | Epstein Barr Virus |
| CMV | Cytomegalovirus |
| Stx11 | Syntaxin 11 |
| StxBP2 | Syntaxin binding protein 2 |
| NVU | Neurovascular unit |
| CSF | Cerebral spinal fluid |
| CEC | Cerebral endothelial cell |
| TMEV | Theiler’s Murine encephalomyelitis virus |
| PIFS | Peptide induced fatal syndrome |
| ECM | Experimental cerebral malaria |
| CM | Cerebral malaria |
| VHF | Viral hemorrhagic fever |
| LCMV | Lymphocytic choriomeningitis virus |
References
- Tschopp, J.; Nabholz, M. Perforin-mediated target cell lysis by cytolytic t lymphocytes. Annu. Rev. Immunol. 1990, 8, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.E.; Kondos, S.C.; Matthews, A.Y.; D’Angelo, M.E.; Dunstone, M.A.; Whisstock, J.C.; Trapani, J.A.; Bird, P.I. The perforin pore facilitates the delivery of cationic cargos. J. Biol. Chem. 2014, 289, 9172–9181. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.A.; Brennan, A.J.; Whisstock, J.C.; Voskoboinik, I.; Trapani, J.A. Protecting a serial killer: Pathways for perforin trafficking and self-defence ensure sequential target cell death. Trends Immunol. 2012, 33, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.A.; Susanto, O.; Jenkins, M.R.; Lukoyanova, N.; Sutton, V.R.; Law, R.H.; Johnston, A.; Bird, C.H.; Bird, P.I.; Whisstock, J.C.; et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013, 121, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Stepp, S.E.; Dufourcq-Lagelouse, R.; Le Deist, F.; Bhawan, S.; Certain, S.; Mathew, P.A.; Henter, J.I.; Bennett, M.; Fischer, A.; de Saint Basile, G.; et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999, 286, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Suidan, G.L.; McDole, J.R.; Chen, Y.; Pirko, I.; Johnson, A.J. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS ONE 2008, 3, e3037. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.M.; Matloubian, M.; Liu, C.C.; Ueda, R.; Kurahara, C.G.; Christensen, J.L.; Huang, M.T.; Young, J.D.; Ahmed, R.; Clark, W.R. Immune function in mice lacking the perforin gene. Proc. Natl. Acad. Sci. USA 1994, 91, 10854–10858. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Hildeman, D.; Kappler, J.; Marrack, P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004, 104, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.; Hien, T.T.; Day, N.; Mai, N.T.; Chuong, L.V.; Chau, T.T.; Loc, P.P.; Phu, N.H.; Bethell, D.; Farrar, J.; et al. Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol. Appl. Neurobiol. 1999, 25, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef] [PubMed]
- McGavern, D.B.; Kang, S.S. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011, 11, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. Vegf-mediated disruption of endothelial cln-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.L.; Chen, Y.; Jin, F.; Hanson, L.M.; Gamez, J.D.; Pirko, I.; Johnson, A.J. Cd8 t cell-initiated blood-brain barrier disruption is independent of neutrophil support. J. Immunol. 2012, 189, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.L.; Willenbring, R.C.; Jin, F.; Manhart, W.A.; LaFrance, S.J.; Pirko, I.; Johnson, A.J. Perforin competent CD8 T cells are sufficient to cause immune-mediated blood-brain barrier disruption. PLoS ONE 2014, 9, e111401. [Google Scholar] [CrossRef] [PubMed]
- Suidan, G.L.; Dickerson, J.W.; Chen, Y.; McDole, J.R.; Tripathi, P.; Pirko, I.; Seroogy, K.B.; Johnson, A.J. Cd8 t cell-initiated vascular endothelial growth factor expression promotes central nervous system vascular permeability under neuroinflammatory conditions. J. Immunol. 2010, 184, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Gloor, S.M.; Wachtel, M.; Bolliger, M.F.; Ishihara, H.; Landmann, R.; Frei, K. Molecular and cellular permeability control at the blood-brain barrier. Brain Res. Rev. 2001, 36, 258–264. [Google Scholar] [CrossRef]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1β regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.E.; Dunstone, M.A.; Whisstock, J.C.; Trapani, J.A.; Bird, P.I. Perforin evolved from a gene duplication of MPEG1, followed by a complex pattern of gene gain and loss within euteleostomi. BMC Evol. Biol. 2012, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Litman, G.W.; Rast, J.P.; Fugmann, S.D. The origins of vertebrate adaptive immunity. Nat. Rev. Immunol. 2010, 10, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Voskoboinik, I. The complex issue of regulating perforin expression. Trends Immunol. 2007, 28, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Pipkin, M.E.; Lieberman, J. Delivering the kiss of death: Progress on understanding how perforin works. Curr. Opin. Immunol. 2007, 19, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Kondos, S.C.; Hatfaludi, T.; Voskoboinik, I.; Trapani, J.A.; Law, R.H.; Whisstock, J.C.; Dunstone, M.A. The structure and function of mammalian membrane-attack complex/perforin-like proteins. Tissue Antigens 2010, 76, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Takio, K.; Okumura, K. Homology of perforin to the ninth component of complement (C9). Nature 1988, 334, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.; Lukoyanova, N.; Voskoboinik, I.; Caradoc-Davies, T.T.; Baran, K.; Dunstone, M.A.; D’Angelo, M.E.; Orlova, E.V.; Coulibaly, F.; Verschoor, S.; et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 2010, 468, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Zagury, D.; Noelle Thierness, J.B.; Feldman, M.; Berke, G. Isolation and characterization of individual functionally reactive cytotoxic t lymphocytes: Conjugation, killing and recycling at teh single cell level. Eur J. Immunol 1975, 5, 818–822. [Google Scholar] [CrossRef]
- Podack, E.R.; Dennert, G. Assembly of two types of tubules with putative cytolytic function by cloned natural killer cells. Nature 1983, 302, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Dourmashkin, R.R.; Deteix, P.; Simone, C.B.; Henkart, P. Electron microscopic demonstration of lesions in target cell membranes associated with antibody-dependent cellular cytotoxicity. Clin. Exp. Immunol. 1980, 42, 554–560. [Google Scholar] [PubMed]
- Kish, D.D.; Gorbachev, A.V.; Parameswaran, N.; Gupta, N.; Fairchild, R.L. Neutrophil expression of fas ligand and perforin directs effector cd8 t cell infiltration into antigen-challenged skin. J. Immunol. 2012, 189, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Knickelbein, J.E.; Ferko, C.; Hendricks, R.L.; Kinchington, P.R. Development and pathogenic evaluation of recombinant herpes simplex virus type 1 expressing two fluorescent reporter genes from different lytic promoters. Virology 2008, 378, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Lykens, J.E.; Terrell, C.E.; Zoller, E.E.; Risma, K.; Jordan, M.B. Perforin is a critical physiologic regulator of T-cell activation. Blood 2011, 118, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Terrell, C.E.; Jordan, M.B. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8+ T cells and dendritic cells. Blood 2013, 121, 5184–5191. [Google Scholar] [CrossRef] [PubMed]
- Clementi, R.; Chiocchetti, A.; Cappellano, G.; Cerutti, E.; Ferretti, M.; Orilieri, E.; Dianzani, I.; Ferrarini, M.; Bregni, M.; Danesino, C.; et al. Variations of the perforin gene in patients with autoimmunity/lymphoproliferation and defective fas function. Blood 2006, 108, 3079–3084. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, J.; Menasche, G.; Callebaut, I.; Minard-Colin, V.; Bader-Meunier, B.; Le Clainche, L.; Fischer, A.; Le Deist, F.; Tardieu, M.; de Saint Basile, G. Severe and progressive encephalitis as a presenting manifestation of a novel missense perforin mutation and impaired cytolytic activity. Blood 2005, 105, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Horne, A.; Ramme, K.G.; Rudd, E.; Zheng, C.; Wali, Y.; al-Lamki, Z.; Gurgey, A.; Yalman, N.; Nordenskjold, M.; Henter, J.I. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br. J. Haematol. 2008, 143, 75–83. [Google Scholar] [CrossRef] [PubMed]
- An, O.; Gursoy, A.; Gurgey, A.; Keskin, O. Structural and functional analysis of perforin mutations in association with clinical data of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) patients. Protein Sci. 2013, 22, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Thia, M.C.; Trapani, J.A. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood 2005, 105, 4700–4706. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Orilieri, E.; Comi, C.; Chiocchetti, A.; Bocca, S.; Boggio, E.; Bernardone, I.S.; Cometa, A.; Clementi, R.; Barizzone, N.; et al. Variations of the perforin gene in patients with multiple sclerosis. Genes Immun. 2008, 9, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Zhang, J.; Wei, Q.; Tang, R.; Qi, J.; Li, L.; Ye, L.; Wang, J. Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood. PLoS ONE 2014, 9, e107386. [Google Scholar] [CrossRef] [PubMed]
- Solomou, E.E.; Gibellini, F.; Stewart, B.; Malide, D.; Berg, M.; Visconte, V.; Green, S.; Childs, R.; Chanock, S.J.; Young, N.S. Perforin gene mutations in patients with acquired aplastic anemia. Blood 2007, 109, 5234–5237. [Google Scholar] [CrossRef] [PubMed]
- Ishii, E.; Ohga, S.; Imashuku, S.; Kimura, N.; Ueda, I.; Morimoto, A.; Yamamoto, K.; Yasukawa, M. Review of hemophagocytic lymphohistiocytosis (HLH) in children with focus on japanese experiences. Crit. Rev. Oncol. Hematol. 2005, 53, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Molleran Lee, S.; Villanueva, J.; Sumegi, J.; Zhang, K.; Kogawa, K.; Davis, J.; Filipovich, A.H. Characterisation of diverse prf1 mutations leading to decreased natural killer cell activity in north american families with haemophagocytic lymphohistiocytosis. J. Med. Genet. 2004, 41, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Muralitharan, S.; Wali, Y.A.; Dennison, D.; Lamki, Z.A.; Zachariah, M.; Nagwa el, B.; Pathare, A.; Krishnamoorthy, R. Novel spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis in ethnic omani patients. Am. J. Hematol. 2007, 82, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, S.; Madkaikar, M.; Desai, M.; Ghosh, K. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis (FHL) patients in india. Blood Cells Mol. Dis. 2015, 54, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Zur Stadt, U.; Beutel, K.; Kolberg, S.; Schneppenheim, R.; Kabisch, H.; Janka, G.; Hennies, H.C. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: Molecular and functional analyses of PRF1, UNC13D, STX11, AND RAB27A. Hum. Mutat. 2006, 27, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Urrea Moreno, R.; Gil, J.; Rodriguez-Sainz, C.; Cela, E.; LaFay, V.; Oloizia, B.; Herr, A.B.; Sumegi, J.; Jordan, M.B.; Risma, K.A. Functional assessment of perforin C2 domain mutations illustrates the critical role for calcium-dependent lipid binding in perforin cytotoxic function. Blood 2009, 113, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Husami, A. Perforin 1 (Pore Forming Protein) (PRF1). Available online: https://research.cchmc.org/LOVD2/ (accession on 11 December 2011).
- Voskoboinik, I.; Sutton, V.R.; Ciccone, A.; House, C.M.; Chia, J.; Darcy, P.K.; Yagita, H.; Trapani, J.A. Perforin activity and immune homeostasis: The common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood 2007, 110, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Risma, K.A.; Frayer, R.W.; Filipovich, A.H.; Sumegi, J. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J. Clin. Investig. 2006, 116, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.J.; Chia, J.; Trapani, J.A.; Voskoboinik, I. Perforin deficiency and susceptibility to cancer. Cell Death Differ 2010, 17, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Dunstone, M.A.; Baran, K.; Whisstock, J.C.; Trapani, J.A. Perforin: Structure, function, and role in human immunopathology. Immunol. Rev. 2010, 235, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Trapani, J.A. Addressing the mysteries of perforin function. Immunol. Cell Biol. 2006, 84, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Trapani, J.A. Perforinopathy: A spectrum of human immune disease caused by defective perforin delivery or function. Front. Immunol. 2013, 4, 441. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Allen, C.E.; Weitzman, S.; Filipovich, A.H.; McClain, K.L. How I treat hemophagocytic lymphohistiocytosis. Blood 2011, 118, 4041–4052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, R.; Wang, J.; Li, L.J.; Song, J.; Qu, W.; Wang, H.Q.; Xing, L.M.; Liu, H.; Wu, Y.H.; et al. Perforin gene mutations in patients with acquired severe aplastic anemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2011, 19, 431–434. [Google Scholar] [PubMed]
- Gholam, C.; Grigoriadou, S.; Gilmour, K.C.; Gaspar, H.B. Familial haemophagocytic lymphohistiocytosis: Advances in the genetic basis, diagnosis and management. Clin. Exp. Immunol. 2011, 163, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Sumegi, J.; Villanueva, J.; Tabata, Y.; Zhang, K.; Chakraborty, R.; Sheng, X.; Clementi, R.; de Saint Basile, G.; Filipovich, A.H. Patients of african ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J. Pediatr. 2006, 149, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Revelo, X.S.; Tsai, S.; Lei, H.; Luck, H.; Ghazarian, M.; Tsui, H.; Shi, S.Y.; Schroer, S.; Luk, C.; Lin, G.H.; et al. Perforin is a novel immune regulator of obesity related insulin resistance. Diabetes 2014, 64, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Risma, K.; Jordan, M.B. Hemophagocytic lymphohistiocytosis: Updates and evolving concepts. Curr. Opin. Pediatr. 2012, 24, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, J.W.; Claireaux, A.E. Familial haemophagocytic reticulosis. Arch. Dis. Child. 1952, 27, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Terrell, C.E.; Jordan, M.B. Mixed hematopoietic or T-cell chimerism above a minimal threshold restores perforin-dependent immune regulation in perforin-deficient mice. Blood 2013, 122, 2618–2621. [Google Scholar] [CrossRef] [PubMed]
- Arico, M.; Danesino, C.; Pende, D.; Moretta, L. Pathogenesis of haemophagocytic lymphohistiocytosis. Br. J. Haematol 2001, 114, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Janka, G.; zur Stadt, U. Familial and acquired hemophagocytic lymphohistiocytosis. Hematol. Am. Soc. Hematol. Educ. Program. 2005, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, J.; Callebaut, I.; Raposo, G.; Certain, S.; Bacq, D.; Dumont, C.; Lambert, N.; Ouachee-Chardin, M.; Chedeville, G.; Tamary, H.; et al. Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003, 115, 461–473. [Google Scholar] [CrossRef]
- zur Stadt, U.; Schmidt, S.; Kasper, B.; Beutel, K.; Diler, A.S.; Henter, J.I.; Kabisch, H.; Schneppenheim, R.; Nurnberg, P.; Janka, G.; et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet. 2005, 14, 827–834. [Google Scholar] [CrossRef] [PubMed]
- zur Stadt, U.; Rohr, J.; Seifert, W.; Koch, F.; Grieve, S.; Pagel, J.; Strauss, J.; Kasper, B.; Nurnberg, G.; Becker, C.; et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in munc18–2 and impaired binding to syntaxin 11. Am. J. Hum. Gene. 2009, 85, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.I.; Elinder, G.; Soder, O.; Ost, A. Incidence in sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr. Scand. 1991, 80, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Donbak, L. Consanguinity in kahramanmaras city, turkey, and its medical impact. Saudi Med. J. 2004, 25, 1991–1994. [Google Scholar] [PubMed]
- Zur Stadt, U.; Beutel, K.; Weber, B.; Kabisch, H.; Schneppenheim, R.; Janka, G. A91V is a polymorphism in the perforin gene not causative of an FHLH phenotype. Blood 2004, 104, 1909–1910. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.N.; Obaro, S.K. Sickle cell disease and malaria morbidity: A tale with two tails. Trends Parasitol. 2011, 27, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Nitcheu, J.; Bonduelle, O.; Combadiere, C.; Tefit, M.; Seilhean, D.; Mazier, D.; Combadiere, B. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J. Immunol. 2003, 170, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia, the first line of defence in brain pathologies. Arzneimittelforschung 1995, 45, 357–360. [Google Scholar] [PubMed]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Brown, K.A. Factors modifying the migration of lymphocytes across the blood-brain barrier. Int. Immunopharmacol. 2001, 1, 2043–2062. [Google Scholar] [CrossRef]
- Mongkolsapaya, J.; Dejnirattisai, W.; Xu, X.N.; Vasanawathana, S.; Tangthawornchaikul, N.; Chairunsri, A.; Sawasdivorn, S.; Duangchinda, T.; Dong, T.; Rowland-Jones, S.; et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat. Med. 2003, 9, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef] [PubMed]
- de Vries, N.A.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Blood-brain barrier and chemotherapeutic treatment of brain tumors. Expert Rev. Neurother. 2006, 6, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, W.N.; Kreidie, M.A.; Kim, R.C.; Hasso, A.N. Acute hemorrhagic leukoencephalitis: Neuroimaging features and neuropathologic diagnosis. J. Comput. Assist. Tomogr. 2005, 29, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Janssen, H.L.; Bienfait, H.P.; Jansen, C.L.; van Duinen, S.G.; Vriesendorp, R.; Schimsheimer, R.J.; Groen, J.; Osterhaus, A.D. Fatal cerebral oedema associated with primary dengue infection. J. Infect. 1998, 36, 344–346. [Google Scholar] [CrossRef]
- Kabakus, N.; Gurgoze, M.K.; Yildirim, H.; Godekmerdan, A.; Aydin, M. Acute hemorrhagic leukoencephalitis manifesting as intracerebral hemorrhage associated with herpes simplex virus type I. J. Trop. Pediatr. 2005, 51, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.J.; Bielefeldt-Ohmann, H. Dengue viral infections; pathogenesis and epidemiology. Microbes Infect. 2000, 2, 1041–1050. [Google Scholar] [CrossRef]
- Medana, I.M.; Turner, G.D. Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 2006, 36, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mul. Scler. 2003, 9, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Pirko, I.; Suidan, G.L.; Rodriguez, M.; Johnson, A.J. Acute hemorrhagic demyelination in a murine model of multiple sclerosis. J. Neuroinflamm. 2008, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Shacklett, B.L.; Cox, C.A.; Wilkens, D.T.; Karl Karlsson, R.; Nilsson, A.; Nixon, D.F.; Price, R.W. Increased adhesion molecule and chemokine receptor expression on cd8+ t cells trafficking to cerebrospinal fluid in HIV-1 infection. J. Infect. Dis. 2004, 189, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Dung, N.M.; Vaughn, D.W.; Kneen, R.; Thao, L.T.; Raengsakulrach, B.; Loan, H.T.; Day, N.P.; Farrar, J.; Myint, K.S.; et al. Neurological manifestations of dengue infection. Lancet 2000, 355, 1053–1059. [Google Scholar] [CrossRef]
- Stoll, G.; Jander, S.; Schroeter, M. Cytokines in cns disorders: Neurotoxicity versus neuroprotection. J. Neural. Transm. Suppl. 2000, 59, 81–89. [Google Scholar] [PubMed]
- Stone, L.A.; Smith, M.E.; Albert, P.S.; Bash, C.N.; Maloni, H.; Frank, J.A.; McFarland, H.F. Blood-brain barrier disruption on contrast-enhanced MRI in patients with mild relapsing-remitting multiple sclerosis: Relationship to course, gender, and age. Neurology 1995, 45, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Talavera, D.; Castillo, A.M.; Dominguez, M.C.; Gutierrez, A.E.; Meza, I. Il8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J Gen. Virol. 2004, 85, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, E.D.; Terajima, M.; Koster, F.T.; Catalina, M.D.; Cruz, J.; Ennis, F.A. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 2004, 172, 3297–3304. [Google Scholar] [CrossRef] [PubMed]
- Paessler, S.; Walker, D.H. Pathogenesis of the viral hemorrhagic fevers. Annu. Rev. Pathol. 2013, 8, 411–440. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.L.; Jin, F.; Pirko, I.; Johnson, A.J. Theiler’s murine encephalomyelitis virus as an experimental model system to study the mechanism of blood-brain barrier disruption. J. Neurovirol. 2013, 20, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Upshaw, J.; Pavelko, K.D.; Rodriguez, M.; Pease, L.R. Preservation of motor function by inhibition of CD8+ virus peptide-specific T cells in theiler’s virus infection. FASEB J. 2001, 15, 2760–2762. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Mendez-Fernandez, Y.; Moyer, A.M.; Sloma, C.R.; Pirko, I.; Block, M.S.; Rodriguez, M.; Pease, L.R. Antigen-specific CD8+ T cells mediate a peptide-induced fatal syndrome. J. Immunol. 2005, 174, 6854–6862. [Google Scholar] [CrossRef] [PubMed]
- Terajima, M.; Ennis, F.A. T cells and pathogenesis of hantavirus cardiopulmonary syndrome and hemorrhagic fever with renal syndrome. Viruses 2011, 3, 1059–1073. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Njenga, M.K.; Hansen, M.J.; Kuhns, S.T.; Chen, L.; Rodriguez, M.; Pease, L.R. Prevalent class i-restricted t-cell response to the theiler’s virus epitope Db:VP2121–130 in the absence of endogenous CD4 help, tumor necrosis factor α, gamma interferon, perforin, or costimulation through CD28. J. Virol. 1999, 73, 3702–3708. [Google Scholar] [PubMed]
- Husami, A. Leiden Open Variation Database Complement Component 9 (C9). Available online: https://research.cchmc.org/LOVD2/home.php?select_db=C9 (accessed on 1 May 2014).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Change | Protein Change | Domain Location | Functional Consequence | Reference |
|---|---|---|---|---|
| 272C>T | A91V | Region of Low homology I | Decreased activity | 38 |
| 548T>G | V183G | Region of Low homology I | Activity maintained | 5 |
| 577T>C | F193L | Region of Low homology I | Conformational change, protein degraded | 52 |
| 662C>T | T221I | Amphipathic α-helix | Conformational change, protein degraded | 52 |
| 673C>T | R225W | Amphipathic α-helix | Activity abolished | 52 |
| 695G>A | R232H | Amphipathic α-helix | Decreased activity | 52 |
| 755A>G | N252S | Region of Low homology II | Activity maintained | 46 |
| 822C>T | A274A | Region of Low homology II | Activity maintained | 46 |
| 836G>A | C279Y | Region of Low homology II | Activity abolished | 5 |
| 900C>T | H300H | Region of Low homology II | Activity maintained | 46 |
| 1122G>A | W374X | Region of Low homology II | Activity abolished | 5 |
| 1228C>T | R410W | EGF like domain | Conformational change, protein degraded | 52 |
| 1229G>C | R410P | EGF like domain | Conformational change, protein degraded | 52 |
| 1286G>A | G429E | C2 Domain | Activity abolished | 52 |
| 1304C>T | T435M | C2 Domain | Activity abolished | 50 |
| 1313A>G | Y438C | C2 Domain | Matures improperly, some activity present | 50 |
| FHL Type | Percentage of FHL Types for All FHL Cases | Gene | Protein Size (Amino Acids) | Number of Mutations in Coding Region of Gene | Mutation Rate (per 100 Amino Accids) |
|---|---|---|---|---|---|
| FHL 1 | 4 cases total | Unknown | Unknown | Unknown | Unknown |
| FHL 2 | 20–40% (>50% in African American families) | PRF1 | 555 | 133 | 23.9 |
| FHL 3 | 20–30% | UNC13D | 1090 | 38 | 3.5 |
| FHL 4 | 20% in Turkish families | STX11 | 287 | 8 | 2.7 |
| FHL 5 | 15–20% | STXBP2 | 593 | 18 | 3.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willenbring, R.C.; Johnson, A.J. Finding a Balance between Protection and Pathology: The Dual Role of Perforin in Human Disease. Int. J. Mol. Sci. 2017, 18, 1608. https://doi.org/10.3390/ijms18081608
Willenbring RC, Johnson AJ. Finding a Balance between Protection and Pathology: The Dual Role of Perforin in Human Disease. International Journal of Molecular Sciences. 2017; 18(8):1608. https://doi.org/10.3390/ijms18081608
Chicago/Turabian StyleWillenbring, Robin C., and Aaron J. Johnson. 2017. "Finding a Balance between Protection and Pathology: The Dual Role of Perforin in Human Disease" International Journal of Molecular Sciences 18, no. 8: 1608. https://doi.org/10.3390/ijms18081608
APA StyleWillenbring, R. C., & Johnson, A. J. (2017). Finding a Balance between Protection and Pathology: The Dual Role of Perforin in Human Disease. International Journal of Molecular Sciences, 18(8), 1608. https://doi.org/10.3390/ijms18081608