Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia?

Abstract
:1. Introduction
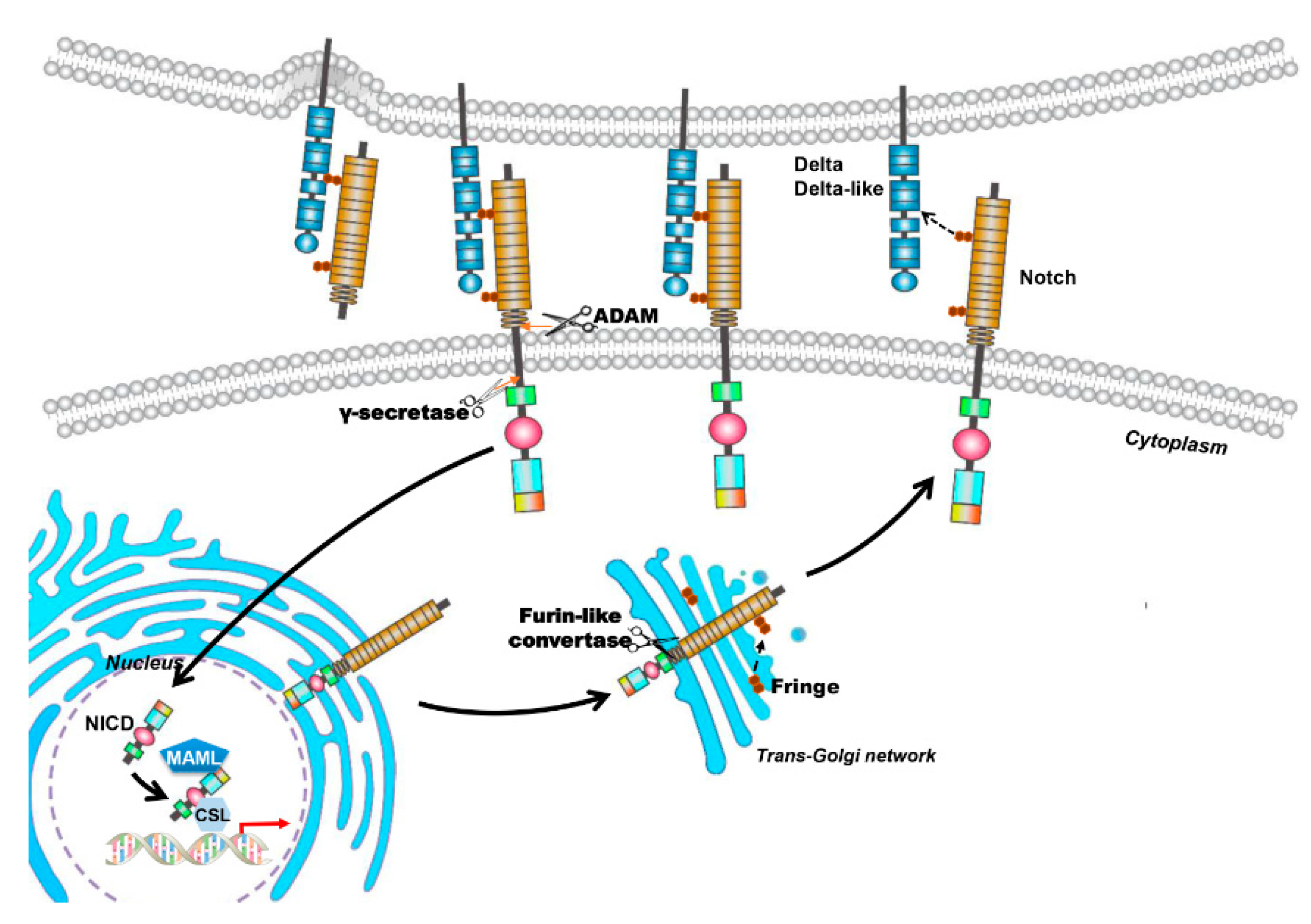
2. The Notch Signaling Pathway
3. Endothelial Cell Proliferation and Regeneration
4. Endothelial Cell Apoptosis
5. Endothelial-Mesenchymal Transition
6. The Contact Interaction between Endothelial Cells and Smooth Muscle Cells
7. Endothelial Cell Secretory Function
8. Endothelial Cell Barrier Dysfunction
9. Limits and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloproteinases |
| AGS | alagille syndrome |
| APOD | apolipoprotein D |
| AVF | arteriovenous fistula |
| AVG | arteriovenous graft |
| bFGF | basic fibroblast growth factor |
| CACs | circulating angiogenic cells |
| CADASIL | cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| CKD | chronic kidney disease |
| CVD | cardiovascular diseases |
| DAPT | N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester |
| Dll | delta-like |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| EndMT | Endothelial-mesenchymal transition |
| eNOS | endothelial NO synthase |
| EPC | endothelial progenitor cell |
| FGFR1 | Fibroblast growth factor receptor1 |
| Frs2α | Fibroblast growth factor receptor substrate 2 α |
| FSP-1 | fibroblast-specific protein 1 |
| HAEC | human arterial endothelial cell |
| HERP | HES-related repressor protein |
| HES | hairy and enhancer of split |
| HUVEC | human umbilical vein endothelial cell |
| IFNγ | Interferon γ |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| MAML | Mastermind-like |
| MAPK | mitogen-activated protein kinase |
| MSC | mesenchymal stem cell |
| NF-κB | nuclear factor κB |
| NICD | Notch intracellular domain |
| NO | Nitric oxide |
| PDGF | platelet-derived growth factor |
| PECAM-1 | platelet endothelial cell adhesion molecule-1 |
| Rb | retinoblastoma |
| ROS | reactive oxygen species |
| α-SMA | α-smooth muscle actin |
| SPARC | secreted protein acidic and rich in cysteine |
| TA | transplant arteriosclerosis |
| TGF-β | transforming growth factor-β |
| TNF | tumor necrosis factor |
| VEGF | vascular endothelial growth factor |
| VSMC | vascular smooth muscle cell |
| WT | wild type |
References
- Brahmbhatt, A.; Remuzzi, A.; Franzoni, M.; Misra, S. The molecular mechanisms of hemodialysis vascular access failure. Kidney Int. 2016, 89, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Zhao, X.J.; Xiang, X.Y. Gene therapy for vein graft failure. J. Card. Surg. 2013, 28, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Ahanchi, S.S.; Tsihlis, N.D.; Kibbe, M.R. The role of nitric oxide in the pathophysiology of intimal hyperplasia. J. Vasc. Surg. 2007, 45, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Butt, M.; Dwivedi, G.; Blann, A.; Khair, O.; Lip, G.Y. Endothelial dysfunction: Methods of assessment & implications for cardiovascular diseases. Curr. Pharm. Des. 2010, 16, 3442–3454. [Google Scholar] [PubMed]
- Allaire, E.; Clowes, A.W. Endothelial cell injury in cardiovascular surgery: The intimal hyperplastic response. Ann. Thorac. Surg. 1997, 63, 582–591. [Google Scholar] [PubMed]
- Bhardwaj, S.; Roy, H.; Heikura, T.; Yla-Herttuala, S. VEGF-A, VEGF-D and VEGF-D (DeltaNDeltaC) induced intimal hyperplasia in carotid arteries. Eur. J. Clin. Investig. 2005, 35, 669–676. [Google Scholar] [CrossRef] [PubMed]
- DiRenzo, D.M.; Chaudhary, M.A.; Shi, X.; Franco, S.R.; Zent, J.; Wang, K.; Guo, L.W.; Kent, K.C. A crosstalk between TGF-β/Smad3 and Wnt/β-catenin pathways promotes vascular smooth muscle cell proliferation. Cell Signal. 2016, 28, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Xu, C.; Yang, J.; Guo, Q.; Hu, Q.; Jiang, H. Resveratrol inhibits phenotypic switching of neointimal vascular smooth muscle cells after balloon injury through blockade of Notch pathway. J. Cardiovasc. Pharmacol. 2014, 63, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Hellstrom, M. Notch as a hub for signaling in angiogenesis. Exp. Cell. Res. 2013, 319, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Gama-Norton, L.; Ferrando, E.; Ruiz-Herguido, C.; Liu, Z.; Guiu, J.; Islam, A.B.; Lee, S.U.; Yan, M.; Guidos, C.J.; Lopez-Bigas, N.; et al. Notch signal strength controls cell fate in the haemogenic endothelium. Nat. Commun. 2015, 6, 8510. [Google Scholar] [CrossRef] [PubMed]
- De Celis, J.F.; Garcia-Bellido, A. Roles of the Notch gene in Drosophila wing morphogenesis. Mech. Dev. 1994, 46, 109–122. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Iruela-Arispe, M.L. Notch signaling in blood vessels: Who is talking to whom about what? Circ. Res. 2007, 100, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Shutter, J.R.; Tanigaki, K.; Honjo, T.; Stark, K.L.; Gridley, T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004, 18, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Arboleda-Velasquez, J.F.; Artavanis-Tsakonas, S. CADASIL: A critical look at a Notch disease. Dev. Neurosci. 2006, 28, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Gridley, T. Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 2003, 12, 9–13. [Google Scholar] [CrossRef]
- Liang, M.; Wang, Y.; Liang, A.; Dong, J.F.; Du, J.; Cheng, J. Impaired integrin β3 delays endothelial cell regeneration and contributes to arteriovenous graft failure in mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Koga, J.; Nakano, T.; Dahlman, J.E.; Figueiredo, J.L.; Zhang, H.; Decano, J.; Khan, O.F.; Niida, T.; Iwata, H.; Aster, J.C.; et al. Macrophage notch ligand delta-like 4 promotes vein graft lesion development: Implications for the treatment of vein graft failure. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2343–2353. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Wang, Y.; Liang, A.; Mitch, W.E.; Roy-Chaudhury, P.; Han, G.; Cheng, J. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires Notch activation to form neointima. Kidney Int. 2015, 88, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Caolo, V.; Schulten, H.M.; Zhuang, Z.W.; Murakami, M.; Wagenaar, A.; Verbruggen, S.; Molin, D.G.; Post, M.J. Soluble Jagged-1 inhibits neointima formation by attenuating Notch-Herp2 signaling. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, A.; Luo, J.; Liang, M.; Han, G.; Mitch, W.E.; Cheng, J. Blocking notch in endothelial cells prevents arteriovenous fistula failure despite CKD. J. Am. Soc. Nephrol. 2014, 25, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; Chang, L.; McLean, G.; Grim, J.E.; Clurman, B.E.; Smith, L.L.; Karsan, A. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: Role of p21Cip1 repression. Mol. Cell Biol. 2004, 24, 8813–8822. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.D.; Ariza-McNaughton, L.; Bermange, A.L.; McAdow, R.; Johnson, S.L.; Lewis, J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 2007, 134, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Sainson, R.C.; Aoto, J.; Nakatsu, M.N.; Holderfield, M.; Conn, E.; Koller, E.; Hughes, C.C. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 2005, 19, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Hobson, B.; Denekamp, J. Endothelial proliferation in tumours and normal tissues: Continuous labelling studies. Br. J. Cancer 1984, 49, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.M.; Wang, S.J.; Taylor, A.C.; Aitkenhead, M.; Hughes, C.C. The basic helix-loop-helix transcription factor HESR1 regulates endothelial cell tube formation. J. Biol. Chem. 2001, 276, 6169–6176. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.L.; Henderson, A.M.; Hughes, C.C. Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvasc. Res. 2002, 64, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Xiao, M.; Balint, K.; Soma, A.; Pinnix, C.C.; Capobianco, A.J.; Velazquez, O.C.; Herlyn, M. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J. 2006, 20, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Trindade, A.; Hirashima, M.; Henrique, D.; Da Costa, L.L.; Rossant, J.; Gill, P.S.; Duarte, A. Loss of Notch signalling induced by Dll4 causes arterial calibre reduction by increasing endothelial cell response to angiogenic stimuli. BMC Dev. Biol. 2008, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Ii, M.; Takeshita, K.; Ibusuki, K.; Luedemann, C.; Wecker, A.; Eaton, E.; Thorne, T.; Asahara, T.; Liao, J.K.; Losordo, D.W. Notch signaling regulates endothelial progenitor cell activity during recovery from arterial injury in hypercholesterolemic mice. Circulation 2010, 121, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Xie, L.; Shi, K.; Zhou, T.; Hua, Y.; Liu, H. Notch signaling change in pulmonary vascular remodeling in rats with pulmonary hypertension and its implication for therapeutic intervention. PLoS ONE 2012, 7, e51514. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.X.; Liang, L.; Wang, L.; Han, J.T.; Zhu, X.X.; Han, H.; Hu, D.H.; Zhang, P. Inhibition of Notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell. Immunol. 2014, 287, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiong, J.; Yang, B.; Zhou, Q.; Wu, Y.; Luo, H.; Zhou, H.; Liu, N.; Li, Y.; Song, Z.; et al. Endothelial cell apoptosis induces TGF-β signaling-dependent host endothelial-mesenchymal transition to promote transplant arteriosclerosis. Am. J. Transplant. 2015, 15, 3095–3111. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Coupel, S.; Coulon, F.; Fitau, J.; Chatelais, M.; Cuturi, M.C.; Chiffoleau, E.; Charreau, B. Impaired Notch4 activity elicits endothelial cell activation and apoptosis: Implication for transplant arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, F.; Duriez, P.; Wong, F.; Noseda, M.; Karsan, A. Notch4 inhibits endothelial apoptosis via RBP-Jkappa-dependent and -independent pathways. J. Biol. Chem. 2004, 279, 11657–11663. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidis, S.; Henze, A.T.; Kruszynska-Ziaja, I.; Skobridis, K.; Theodorou, V.; Paleolog, E.M.; Mazzone, M. Factor-inhibiting HIF-1 (FIH-1) is required for human vascular endothelial cell survival. FASEB J. 2015, 29, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Devalliere, J.; Chatelais, M.; Coulon, F.; Seveno, C.; Romagnoli, M.; Barille Nion, S.; Charreau, B. Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS ONE 2009, 4, e8244. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wei, C.; Kim, I.K.; Janssen-Heininger, Y.; Gupta, S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 2014, 63, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Du, B.; Xie, F.; Cai, W.; Liu, Y.; Li, Y.; Feng, L.; Qiu, L. Vaccarin attenuates high glucose-induced human EA*hy926 endothelial cell injury through inhibition of Notch signaling. Mol. Med. Rep. 2016, 13, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wang, N.; Zhang, T.C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 2012, 64, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Hong, M.S.; Fu, C.; Schmit, B.M.; Su, Y.; Berceli, S.A.; Jiang, Z. Preexisting smooth muscle cells contribute to neointimal cell repopulation at an incidence varying widely among individual lesions. Surgery 2016, 159, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Moonen, J.R.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; Van Kooten, T.G.; Van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor β signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dong, F.; Jia, Y.; Du, H.; Dong, N.; Xu, Y.; Wang, S.; Wu, H.; Liu, Z.; Li, W. Notch signal regulates corneal endothelial-to-mesenchymal transition. Am. J. Pathol. 2013, 183, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Kapela, A.; Tran, C.H.; Welsh, D.G.; Tsoukias, N.M. Role of microprojections in myoendothelial feedback—A theoretical study. J. Physiol. 2013, 591, 2795–2812. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Haddock, R.E.; Hill, C.E.; Chadha, P.S.; Kerr, P.M.; Welsh, D.G.; Plane, F. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin. Exp. Pharmacol. Physiol. 2009, 36, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rostama, B.; Peterson, S.M.; Vary, C.P.; Liaw, L. Notch signal integration in the vasculature during remodeling. Vascul. Pharmacol. 2014, 63, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Scheppke, L.; Murphy, E.A.; Zarpellon, A.; Hofmann, J.J.; Merkulova, A.; Shields, D.J.; Weis, S.M.; Byzova, T.V.; Ruggeri, Z.M.; Iruela-Arispe, M.L.; et al. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood 2012, 119, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.A.; West, X.Z.; Kim, Y.W.; Zhao, Y.; Tischenko, M.; Cull, R.M.; Phares, T.W.; Peng, X.D.; Bernier-Latmani, J.; Petrova, T.V.; et al. Stability and function of adult vasculature is sustained by Akt/Jagged1 signalling axis in endothelium. Nat. Commun. 2016, 7, 10960. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Lin, S.; Sandig, M.; Mequanint, K. Regulation of vascular smooth muscle cell phenotype in three-dimensional coculture system by Jagged1-selective Notch3 signaling. Tissue Eng. Part A 2014, 20, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kennard, S.; Lilly, B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 2009, 104, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Bhattacharyya, A.; Roszell, E.E.; Sandig, M.; Mequanint, K. The role of endothelial cell-bound Jagged1 in Notch3-induced human coronary artery smooth muscle cell differentiation. Biomaterials 2012, 33, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Lilly, B. Notch signaling governs phenotypic modulation of smooth muscle cells. Vascul. Pharmacol. 2014, 63, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xiao, Y.; Mao, Z.; Huang, J.; Geng, Q.; Wang, W.; Dong, P. Soluble Jagged-1 inhibits restenosis of vein graft by attenuating Notch signaling. Microvasc. Res. 2015, 100, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Proweller, A. Vascular smooth muscle Notch signals regulate endothelial cell sensitivity to angiogenic stimulation. J. Biol. Chem. 2011, 286, 13741–13753. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Huang, L.; Fang, Y.; Guo, R.; Yin, Y.; Zhao, X. Transplantation of endothelial progenitor cells overexpressing endothelial nitric oxide synthase enhances inhibition of neointimal hyperplasia and restores endothelium-dependent vasodilatation. Microvasc. Res. 2011, 81, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Tsihlis, N.D.; Oustwani, C.S.; Vavra, A.K.; Jiang, Q.; Keefer, L.K.; Kibbe, M.R. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem. Biophys. 2011, 60, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, M.L.; Heerkens, J.L.; De Bruin, A.M.; Klous, A.; De Waard, V.; De Vries, C.J. Erythropoietin accelerates smooth muscle cell-rich vascular lesion formation in mice through endothelial cell activation involving enhanced PDGF-BB release. Blood 2010, 115, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Gaetano, C.; Antonini, A.; Pompilio, G.; Bracco, E.; Ronnstrand, L.; Heldin, C.H.; Capogrossi, M.C. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: Consequences for smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Deaton, R.A.; Hastings, N.E.; Shang, Y.; Moehle, C.W.; Eriksson, U.; Topouzis, S.; Wamhoff, B.R.; Blackman, B.R.; Owens, G.K. PDGF-DD, a novel mediator of smooth muscle cell phenotypic modulation, is upregulated in endothelial cells exposed to atherosclerosis-prone flow patterns. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Sharghi-Namini, S.; Tan, E.; Ong, L.L.; Ge, R.; Asada, H.H. Dll4-containing exosomes induce capillary sprout retraction in a 3D microenvironment. Sci. Rep. 2014, 4, 4031. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M. The endothelium: Part 1: Multiple functions of the endothelial cells-focus on endothelium-derived vasoactive mediators. In Integrated Systems Physiology: From Molecule to Function to Disease; Granger, D.N., Granger, J.P., Eds.; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011. [Google Scholar]
- Triggle, C.R.; Samuel, S.M.; Ravishankar, S.; Marei, I.; Arunachalam, G.; Ding, H. The endothelium: Influencing vascular smooth muscle in many ways. Can. J. Physiol. Pharmacol. 2012, 90, 713–738. [Google Scholar] [CrossRef] [PubMed]
- Peiro, C.; Redondo, J.; Rodriguez-Martinez, M.A.; Angulo, J.; Marin, J.; Sanchez-Ferrer, C.F. Influence of endothelium on cultured vascular smooth muscle cell proliferation. Hypertension 1995, 25, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E. The role of pericytes in angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Isenberg, J.S.; Roberts, D.D. Molecular regulation of tumor angiogenesis and perfusion via redox signaling. Chem. Rev. 2009, 109, 3099–3124. [Google Scholar] [CrossRef] [PubMed]
- Lavin, B.; Gomez, M.; Pello, O.M.; Castejon, B.; Piedras, M.J.; Saura, M.; Zaragoza, C. Nitric oxide prevents aortic neointimal hyperplasia by controlling macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Bahnson, E.S.; Koo, N.; Cantu-Medellin, N.; Tsui, A.Y.; Havelka, G.E.; Vercammen, J.M.; Jiang, Q.; Kelley, E.E.; Kibbe, M.R. Nitric oxide inhibits neointimal hyperplasia following vascular injury via differential, cell-specific modulation of SOD-1 in the arterial wall. Nitric Oxide 2015, 44, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.G.; Najjar, S.F.; Kapadia, M.R.; Murar, J.; Eng, J.; Lyle, B.; Aalami, O.O.; Jiang, Q.; Hrabie, J.A.; Saavedra, J.E.; et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free Radic. Biol. Med. 2008, 44, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, A.; Fuller, M.; Chang, L.; Wong, F.; Paliouras, G.; Shaw, R.; Kyle, A.H.; Umlandt, P.; Baker, J.H.; Diaz, E.; et al. Endothelial-specific Notch blockade inhibits vascular function and tumor growth through an eNOS-dependent mechanism. Cancer Res. 2014, 74, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Muto, A.; Kudo, F.A.; Model, L.; Eghbalieh, S.; Chowdhary, P.; Dardik, A. Age-related Notch-4 quiescence is associated with altered wall remodeling during vein graft adaptation. J. Surg. Res. 2011, 171, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; McRobb, L.S.; Khachigian, L.M. Inhibition of intimal thickening after vascular injury with a cocktail of vascular endothelial growth factor and cyclic Arg-Gly-Asp peptide. Int. J. Cardiol. 2016, 220, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, D.; Yang, Y.; Ji, K.; Gao, P. Endotheliallike cells differentiated from mesenchymal stem cells attenuate neointimal hyperplasia after vascular injury. Mol. Med. Rep. 2016, 14, 4830–4836. [Google Scholar] [PubMed]
- Janardhanan, R.; Yang, B.; Kilari, S.; Leof, E.B.; Mukhopadhyay, D.; Misra, S. The Role of Repeat Administration of Adventitial Delivery of Lentivirus-shRNA-Vegf-A in Arteriovenous Fistula to Prevent Venous Stenosis Formation. J. Vasc. Interv. Radiol. 2016, 27, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Cheng, G.; Zhou, Y.; Xu, G. Thymosin β4 induces angiogenesis through Notch signaling in endothelial cells. Mol. Cell. Biochem. 2013, 381, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.P.; Wan, A.; Lal, N.; Zhang, D.; Wang, F.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Cardiomyocyte VEGF Regulates Endothelial Cell GPIHBP1 to relocate lipoprotein lipase to the coronary lumen during diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Mouillesseaux, K.P.; Bautch, V.L. Flt-1 (vascular endothelial growth factor receptor-1) is essential for the vascular endothelial growth factor-Notch feedback loop during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Duan, M.; Hu, G.; Buch, S. Platelet-derived growth factor B chain is a novel target gene of cocaine-mediated Notch1 signaling: Implications for HIV-associated neurological disorders. J. Neurosci. 2011, 31, 12449–12454. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.; Czupalla, C.J.; Ziegler, N.; Devraj, K.; Zinke, J.; Seidel, S.; Heck, R.; Thom, S.; Macas, J.; Bockamp, E.; et al. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012, 209, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, B.; Bhoopathi, P.; Chetty, C.; Gogineni, V.R.; Sailaja, G.S.; Gondi, C.S.; Rao, J.S. Notch signaling regulates tumor-induced angiogenesis in SPARC-overexpressed neuroblastoma. Angiogenesis 2013, 16, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Soulez, M.; Sirois, I.; Brassard, N.; Raymond, M.A.; Nicodeme, F.; Noiseux, N.; Durocher, Y.; Pshezhetsky, A.V.; Hebert, M.J. Epidermal growth factor and perlecan fragments produced by apoptotic endothelial cells co-ordinately activate ERK1/2-dependent antiapoptotic pathways in mesenchymal stem cells. Stem Cells 2010, 28, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Pajaniappan, M.; Glober, N.K.; Kennard, S.; Liu, H.; Zhao, N.; Lilly, B. Endothelial cells downregulate apolipoprotein D expression in mural cells through paracrine secretion and Notch signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, A.; Ene-Iordache, B. Novel paradigms for dialysis vascular access: Upstream hemodynamics and vascular remodeling in dialysis access stenosis. Clin. J. Am. Soc. Nephrol. 2013, 8, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Tesfamariam, B. Endothelial Repair and Regeneration Following Intimal Injury. J. Cardiovasc. Transl. Res. 2016, 9, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Tan, Y.; Beecham, G.W.; Seo, D.M.; Tian, R.; Li, Y.; Vazquez-Padron, R.I.; Pericak-Vance, M.; Vance, J.M.; Goldschmidt-Clermont, P.J.; et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012, 225, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Devalliere, J.; Coupel, S.; Charreau, B. Inflammation dysregulates Notch signaling in endothelial cells: Implication of Notch2 and Notch4 to endothelial dysfunction. Biochem. Pharmacol. 2010, 80, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.; Connolly, M.; Gao, W.; McCormick, J.; Biniecka, M.; Sullivan, O.; Kirby, B.; Sweeney, C.; Molloy, E.; Markham, T.; et al. Notch-1 mediates endothelial cell activation and invasion in psoriasis. Exp. Dermatol. 2014, 23, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, S.; Li, K.; Li, L.; Xu, C.; Xiang, B. Signaling pathways in the development of infantile hemangioma. J. Hematol. Oncol. 2014, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Schneider, D.J.; Sobel, B.E.; Dauerman, H.L. Endothelial dysfunction and inflammation after percutaneous coronary intervention. Am. J. Cardiol. 2004, 94, 1420–1423. [Google Scholar] [CrossRef] [PubMed]
- Hofma, S.H.; Van der Giessen, W.J.; Van Dalen, B.M.; Lemos, P.A.; McFadden, E.P.; Sianos, G.; Ligthart, J.M.; Van Essen, D.; De Feyter, P.J.; Serruys, P.W. Indication of long-term endothelial dysfunction after sirolimus-eluting stent implantation. Eur. Heart J. 2006, 27, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Togni, M.; Windecker, S.; Cocchia, R.; Wenaweser, P.; Cook, S.; Billinger, M.; Meier, B.; Hess, O.M. Sirolimus-eluting stents associated with paradoxic coronary vasoconstriction. J. Am. Coll. Cardiol. 2005, 46, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Huang, F.; Yi, Y.; Yin, L.; Peng, D. EGCG attenuates atherosclerosis through the Jagged-1/Notch pathway. Int. J. Mol. Med. 2016, 37, 398–406. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. The Direct Contact between ECs and VSMCs Mediated by Notch | ||||
| Ways of Interaction | Ligand | Receptor | Function | Possible Effect on Neointimal Hyperplasia |
| Direct Contact | ND | VSMC Notch-1 | VSMC migration [64]↑ | Promotion |
| EC Jagged-1 | VSMC Notch-2 | VSMC differentiation [63]↑ | ND | |
| VSMC proliferation [63]↓ | ||||
| EC Jagged-1 | VSMC Notch-3 | VSMC differentiation [60,61,62,63]↑ | Promotion | |
| VSMC secretion [63]↑ | ||||
| VSMC migration [64]↑ | ||||
| VSMC Jagged-1 | EC Notch-1 | EC proliferation [65]↑ | Inhibition | |
| ND | EC Notch-2, -3, -4 [15] | ND | ND | |
| B. The Indirect Communication between ECs and VSMCs Mediated by Notch Activation | ||||
| Ways of Interaction | Notch Signaling | Molecules Secreted from EC | Function | Possible Effect on Neointimal Hyperplasia |
| Indirect Communication | Activation | NO | EC proliferation [66,67]↑ | Inhibition |
| VSMC proliferation [68]↓ | ||||
| VSMC migration [68]↓ | ||||
| VEGF | EC proliferation↑ | Inhibition | ||
| EC regeneration↑ | ||||
| VSMC proliferation↓ | ||||
| PDGF | VSMC proliferation [69]↑ | Promotion | ||
| VSMC migration [70]↑ | ||||
| VSMC differentiation [71]↑ | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, D.-Y.; Jin, X.-R.; Zeng, X.; Wang, Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. https://doi.org/10.3390/ijms18081615
Tian D-Y, Jin X-R, Zeng X, Wang Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? International Journal of Molecular Sciences. 2017; 18(8):1615. https://doi.org/10.3390/ijms18081615
Chicago/Turabian StyleTian, Ding-Yuan, Xu-Rui Jin, Xi Zeng, and Yun Wang. 2017. "Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia?" International Journal of Molecular Sciences 18, no. 8: 1615. https://doi.org/10.3390/ijms18081615
APA StyleTian, D. -Y., Jin, X. -R., Zeng, X., & Wang, Y. (2017). Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? International Journal of Molecular Sciences, 18(8), 1615. https://doi.org/10.3390/ijms18081615