Transcriptome Profiling in Human Diseases: New Advances and Perspectives

 ,
,  ,
, 
 and
and 
Abstract
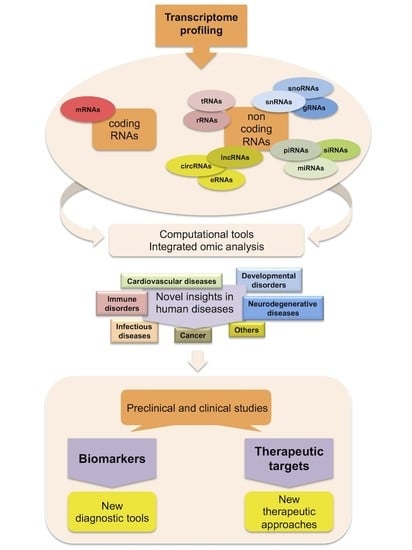
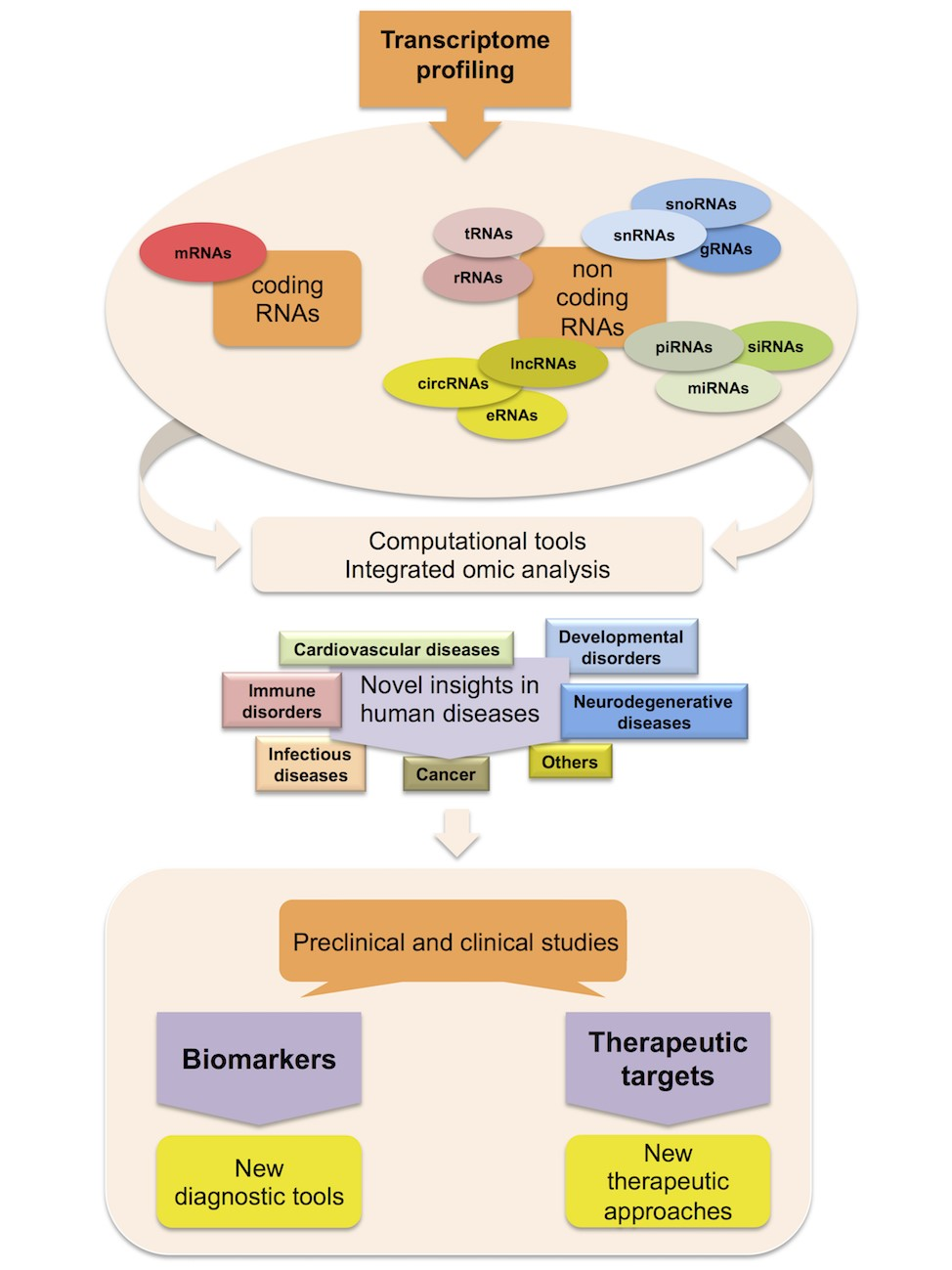
:
{kind=link}
{kind=link}
1. Introduction
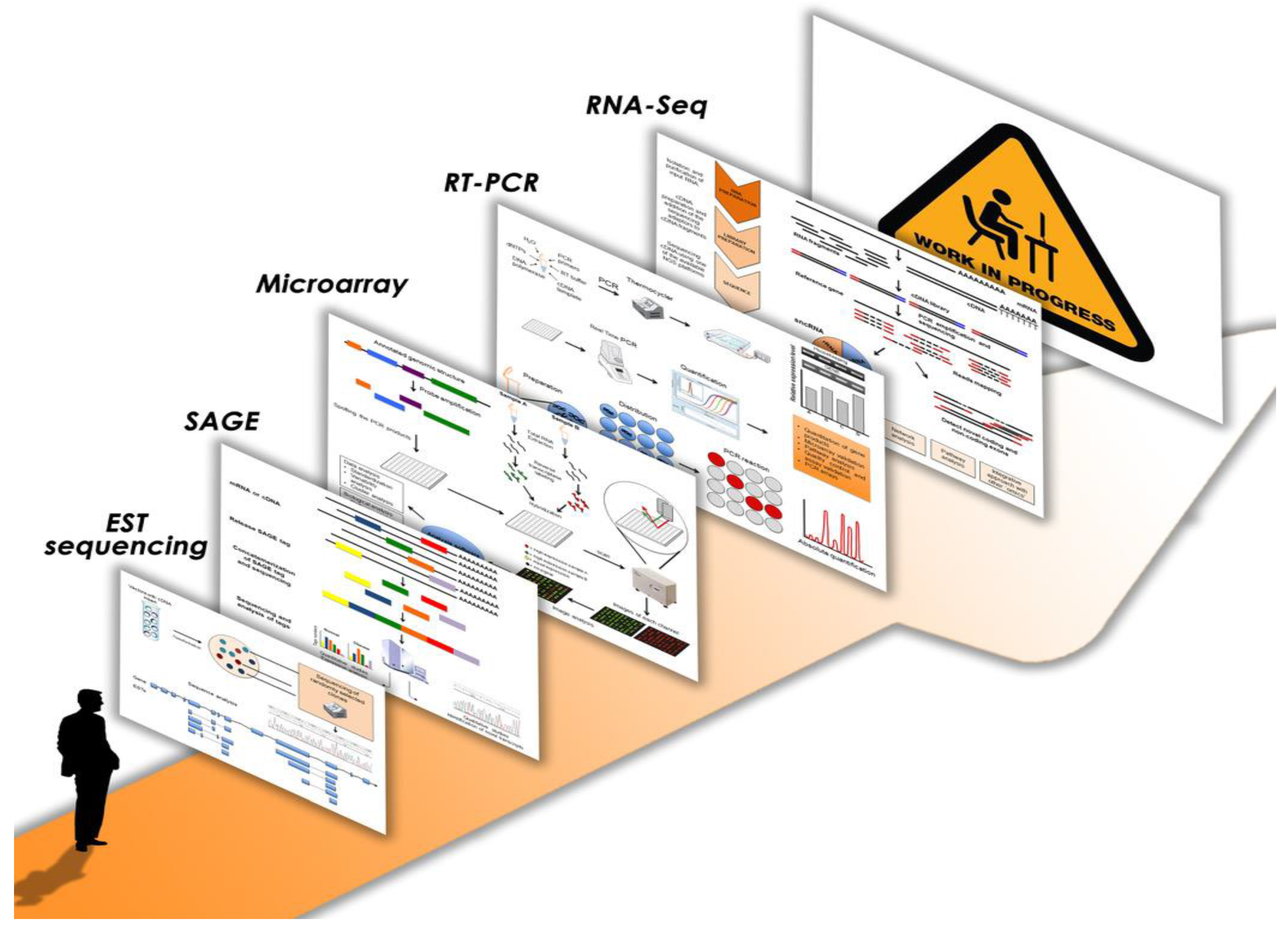
2. Novel Insights from Transcriptome Studies by Next Generation Sequencing
3. Transcriptome Studies by NGS: the Work in Progress
4. Integrated Omic Analysis: Beyond Transcriptomics
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CCA | Canonical Correlation Analysis |
| circRNAs | Circular RNAs |
| dPCR | Digital Polymerase Chain Reaction |
| ENCODE | Encyclopedia of the regulatory elements |
| ESTs | Expressed Sequence Tags |
| GEO | Gene Expression Omnibus |
| gRNAs | Guide RNAs |
| GTEx | Genotype-Tissue Expression |
| ICGC | International Cancer Genome Consortium |
| lncRNAs | Long ncRNAs |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| miRNAs | microRNAs |
| ncRNAs | Non-coding RNAs |
| NGS | Next Generation Sequencing |
| PCA | Principal Component Analysis |
| piRNAs | Piwi-interacting RNAs |
| PLS | Partial Least Square |
| qRT-PCR | Quantitative Reverse Transcription PCR |
| RISC | RNA-induced silencing complex |
| RNA-Seq | RNA-Sequencing |
| SAGE | Serial Analysis of Gene Expression |
| siRNAs | Short interfering RNAs |
| snoRNAs | Small nucleolar RNAs |
| snRNAs | Small nuclear RNAs |
| TCGA | The Cancer Genome Atlas |
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howel, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, A. The complex eukaryotic transcriptome: Unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 2009, 10, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F.; et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef]
- De Risi, J.; Penland, L.; Brown, P.O.; Bittner, M.L.; Meltzer, P.S.; Ray, M.; Chen, Y.; Su, Y.A.; Trent, J.M. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 1996, 14, 457–460. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Hata, R.; Masumura, M.; Akatsu, H.; Li, F.; Fujita, H.; Nagai, Y.; Yamamoto, T.; Okada, H.; Kosaka, K.; Sakanaka, M.; Sawada, T. Up-regulation of calcineurin Aβ mRNA in the Alzheimer’s disease brain: assessment by cDNA microarray. Biochem. Biophys. Res. Commun. 2001, 284, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Whitney, L.W.; Becker, K.G.; Tresser, N.J.; Caballero-Ramos, C.I.; Munson, P.J.; Prabhu, V.V.; Trent, J.M.; McFarland, H.F.; Biddison, W.E. Analysis of gene expression in multiple sclerosis lesions using cDNA microarrays. Ann. Neurol. 1999, 46, 425–428. [Google Scholar] [CrossRef]
- Heller, R.A.; Schena, M.; Chai, A.; Shalon, D.; Bedilion, T.; Gilmore, J.; Woolley, D.E.; Davis, R.W. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc. Natl. Acad. Sci. USA 1997, 94, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Barrans, J.D.; Allen, P.D.; Stamatiou, D.; Dzau, V.J.; Liew, C.C. Global gene expression profiling of end-stage dilated cardiomyopathy using a human cardiovascular-based cDNA microarray. Am. J. Pathol. 2002, 160, 2035–2043. [Google Scholar] [CrossRef]
- Malone, J.H.; Oliver, B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Rienzo, M.; Nagel, J.; Casamassimi, A.; Giovane, A.; Dietzel, S.; Napoli, C. Mediator subunits: Gene expression pattern, a novel transcript identification and nuclear localization in human endothelial progenitor cells. Biochim. Biophys. Acta 2010, 1799, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Rienzo, M.; Casamassimi, A.; Schiano, C.; Grimaldi, V.; Infante, T.; Napoli, C. Distinct alternative splicing patterns of mediator subunit genes during endothelial progenitor cell differentiation. Biochimie 2012, 94, 1828–1832. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Mueller, R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin. Sci. (Lond.) 2005, 109, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Bustin, S.A. Reliability of real-time reverse-transcription PCR in clinical diagnostics: Gold standard or substandard? Expert Rev. Mol. Diagn. 2009, 9, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Cui, X.; Hu, J.; Li, Z.; Choi, J.R.; Yang, Q.; Lin, M.; Ying Hui, L.; Xu, F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2017, 90, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Piccart, M.J. Taking gene-expression profiling to the clinic: When will molecular signatures become relevant to patient care? Nat. Rev. Cancer 2007, 7, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Greco, F.A.; Oien, K.; Erlander, M.; Osborne, R.; Varadhachary, G.; Bridgewater, J.; Cohen, D.; Wasan, H. Cancer of unknown primary: Progress in the search for improved and rapid diagnosis leading toward superior patient outcomes. Ann. Oncol. 2012, 23, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Chibon, F. Cancer gene expression signatures—The rise and fall? Eur. J. Cancer 2013, 49, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Angelini, C.; D’Apice, L.; Mutarelli, M.; Casamassimi, A.; Sommese, L.; Gallo, M.A.; Aprile, M.; Esposito, R.; Leone, L.; et al. Massive-scale RNA-Seq analysis of non-ribosomal transcriptome in human trisomy 21. PLoS ONE 2011, 6, e18493. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Angelini, C.; de Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010, 853916. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Aprile, M.; Esposito, R.; Ciccodicola, A. RNA-Seq and human complex diseases: Recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Scarpato, M.; Federico, A.; Ciccodicola, A.; Costa, V. Novel transcription factor variants through RNA-sequencing: The importance of being “alternative”. Int. J. Mol. Sci. 2015, 16, 1755–1771. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Casamassimi, A.; Peluso, G.; Barone, M.V.; Capaccio, D.; Migliore, C.; Bonelli, P.; Pedicini, A.; Febbraro, A.; Ciccodicola, A.; et al. A novel peroxisome proliferator-activated receptor gamma isoform with dominant negative activity generated by alternative splicing. J. Biol. Chem. 2005, 280, 26517–26525. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Suñé-Pou, M.; Prieto-Sánchez, S.; Boyero-Corral, S.; Moreno-Castro, C.; El Yousfi, Y.; Suñé-Negre, J.M.; Hernández-Munain, C.; Suñé, C. Targeting splicing in the treatment of human disease. Genes 2017, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Park, J.W.; Ramachandran, S.; Zhang, Y.; Tseng, Y.T.; Shen, S.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.; Troncoso, J.C.; et al. Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 3454–3466. [Google Scholar] [CrossRef] [PubMed]
- Kõks, S.; Keermann, M.; Reimann, E.; Prans, E.; Abram, K.; Silm, H.; Kõks, G.; Kingo, K. Psoriasis-Specific RNA Isoforms Identified by RNA-Seq Analysis of 173,446 Transcripts. Front. Med. 2016, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Aversa, R.; Sorrentino, A.; Esposito, R.; Ambrosio, M.R.; Amato, A.; Zambelli, A.; Ciccodicola, A.; D’Apice, L.; Costa, V. Alternative Splicing in Adhesion and Motility-Related Genes in Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Singh, S.; Shukla, S. A saga of cancer epigenetics: Linking epigenetics to alternative splicing. Biochem. J. 2017, 474, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Esposito, R.; Ziviello, C.; Sepe, R.; Bim, L.V.; Cacciola, N.A.; Decaussin-Petrucci, M.; Pallante, P.; Fusco, A.; Ciccodicola, A. New somatic mutations and WNK1-B4GALNT3 gene fusion in papillary thyroid carcinoma. Oncotarget 2015, 6, 11242–11251. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Hayashizaki, Y. Noncoding RNA transcription beyond annotated genes. Curr. Opin. Genet. Dev. 2007, 17, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.A.; Kugel, J.F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol. 2006, 7, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Stroynowska-Czerwinska, A.; Fiszer, A.; Krzyzosiak, W.J. The panorama of miRNA-mediated mechanisms in mammalian cells. Cell Mol. Life Sci. 2014, 71, 2253–2270. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, P.A.; Madhani, H.D. Recognizing the enemy within: licensing RNA-guided genome defense. Trends Biochem. Sci. 2014, 39, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H. PIWI-interacting RNA: Its biogenesis and functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef] [PubMed]
- Khanduja, J.S.; Calvo, I.A.; Joh, R.I.; Hill, I.T.; Motamedi, M. Nuclear noncoding RNAs and genome stability. Mol. Cell 2016, 63, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Tuna, M.; Machado, A.S.; Calin, G.A. Genetic and epigenetic alterations of microRNAs and implications for human cancers and other diseases. Genes Chromosomes Cancer 2016, 55, 193–214. [Google Scholar] [CrossRef] [PubMed]
- miRBase. Available online: http://microrna.sanger.ac.uk (accessed on 5 June 2017).
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Esposito, R.; Aprile, M.; Ciccodicola, A. Non-coding RNA and pseudogenes in neurodegenerative diseases: “The (un)Usual Suspects”. Front. Genet. 2012, 3, 231. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, W.; Wang, J.; Wang, K.; Li, P. The role and molecular mechanism of non-coding RNAs in pathological cardiac remodeling. Int. J. Mol. Sci. 2017, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, J.H.; Spector, D.L. Long non-coding RNAs: Modulators of nuclear structure and function. Curr. Opin. Cell Biol. 2014, 26, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Gloss, B.S.; Dinger, M.E. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta 2016, 1859, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.B.; Mercer, T.R.; Bussotti, G.; Leonardi, T.; Haynes, K.R.; Crawford, J.; Brunck, M.E.; Cao, K.A.; Thomas, G.P.; Chen, W.Y.; et al. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat. Methods 2015, 12, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Gordon, W.; Quan, J.; Xi, H.; Du, S.; von Schack, D.; Zhang, B. Comparison of stranded and non-stranded RNA-seq transcriptome profiling and investigation of gene overlap. BMC Genom. 2015, 16, 675. [Google Scholar] [CrossRef] [PubMed]
- Corley, S.M.; MacKenzie, K.L.; Beverdam, A.; Roddam, L.F.; Wilkins, M.R. Differentially expressed genes from RNA-Seq and functional enrichment results are affected by the choice of single-end versus paired-end reads and stranded versus non-stranded protocols. BMC Genom. 2017, 18, 399. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Fernandez-Jimenez, N.; Kratchmarov, R.; Luo, X.; Bhagat, G.; Green, P.H.; Schneider, R.; Kiledjian, M.; Bilbao, J.R.; Ghosh, S. A long noncoding RNA associated with susceptibility to celiac disease. Science 2016, 352, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, K.K.; Kjems, J.; Hansen, T.B. Circular RNAs: Identification, biogenesis and function. Biochim. Biophys. Acta 2016, 1859, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Chen, T.; Yao, Q.; Zheng, L.; Zhang, Z.; Wang, J.; Hu, Z.; Cui, H.; Han, Y.; Han, X.; Zhang, K.; Hong, W. The circular RNA ciRS-7 promotes APP and BACE1 degradation in an NF-κB-dependent manner. FEBS J. 2017, 284, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Legnini, I.; di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell. 2017, 66, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tsukahara, T. A view of pre-mRNA splicing from RNase R resistant RNAs. Int. J. Mol. Sci. 2014, 15, 9331–9342. [Google Scholar] [CrossRef]
- Qu, S.; Zhong, Y.; Shang, R.; Zhang, X.; Song, W.; Kjems, J.; Li, H. The emerging landscape of circular RNA in life processes. RNA Biol. 2016, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, C.; Tan, C.; Liu, X. Circular RNAs: A new frontier in the study of human diseases. J. Med. Genet. 2016, 53, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.I.; Altman, N. Points of significance: Power and sample size. Nat. Methods 2013, 12, 1139–1140. [Google Scholar] [CrossRef]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, P.; Steijger, T.; Sipos, B.; Grant, G.; Kahles, A.; Rätsch, G.; Goldman, N.; Hubbard, T.; Harrow, J.; Guigo, R.; et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 2017, 541, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Poirion, O.B.; Zhu, X.; Ching, T.; Garmire, L. Single-cell transcriptomics bioinformatics and computational challenges. Front. Genet. 2016, 7, 163. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas. Available online: http://cancergenome.nih.gov (accessed on 5 June 2017).
- ICGC Cancer Genome Projects. Available online: http://icgc.org (accessed on 5 June 2017).
- The Allen Human Brain Atlas. Available online: http://human.brain-map (accessed on 5 June 2017).
- Gene Expression Omnibus (GEO). Available online: http://www.ncbi.nlm.nih.gov/gds (accessed on 5 June 2017).
- ENCODE: Encyclopedia of DNA Elements. Available online: https://www.encodeproject.org (accessed on 5 June 2017).
- The Genotype-Tissue Expression (GTEx) Project. Available online: https://www.gtexportal.org (accessed on 5 June 2017).
- Federico, A.; Rienzo, M.; Abbondanza, C.; Costa, V.; Ciccodicola, A.; Casamassimi, A. Pan-cancer mutational and transcriptional analysis of the integrator complex. Int. J. Mol. Sci. 2017, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Kang, J.; Kim, D.; Kim, K.P.; Ryoo, B.Y.; Hong, S.M.; Hwang, J.J.; Jeong, S.Y.; Hwang, S.; Kim, K.H.; et al. Multiplexed gene expression profiling identifies the FGFR4 pathway as a novel biomarker in intrahepatic cholangiocarcinoma. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, A.; Occhipinti, A.; Angelini, C.; de Feis, I.; Lió, P. Cancer markers selection using network-based Cox regression: A methodological and computational practice. Front. Physiol. 2016, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrero, D.; Abugessaisa, I.; Maier, D.; Teschendorff, A.; Merkenschlager, M.; Gisel, A.; Ballestar, E.; Bongcam-Rudloff, E.; Conesa, A.; Tegnér, J. Data integration in the era of omics: Current and future challenges. BMC Syst. Biol. 2014, 8, I1. [Google Scholar] [CrossRef] [PubMed]
- Lê Cao, K.A.; González, I.; Déjean, S. IntegrOmics: An R package to unravel relationships between two omics datasets. Bioinformatics 2009, 25, 2855–2856. [Google Scholar] [CrossRef] [PubMed]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Akavia, U.D.; Litvin, O.; Kim, J.; Sanchez-Garcia, F.; Kotliar, D.; Causton, H.C.; Pochanard, P.; Mozes, E.; Garraway, L.A.; Pe’er, D. An integrated approach to uncover drivers of cancer. Cell 2010, 143, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- González, I.; Cao, K.A.; Davis, M.J.; Déjean, S. Visualising associations between paired “omics” data sets. BioData Min. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, O.; Mazza, T.; Castellana, S.; Panza, A.; Latiano, T.; Corritore, G.; Andriulli, A.; Latiano, A. Inflammatory bowel disease meets systems biology: A multi-omics challenge and frontier. OMICS 2016, 20, 692–698. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 5 June 2017).
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int. J. Mol. Sci. 2017, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Turner, A.; Aggarwal, P.; Matter, A.; Storvick, E.; Arnett, D.K.; Broeckel, U. Comprehensive evaluation of AmpliSeq transcriptome, a novel targeted whole transcriptome RNA sequencing methodology for global gene expression analysis. BMC Genom. 2015, 16, 1069. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. https://doi.org/10.3390/ijms18081652
Casamassimi A, Federico A, Rienzo M, Esposito S, Ciccodicola A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. International Journal of Molecular Sciences. 2017; 18(8):1652. https://doi.org/10.3390/ijms18081652
Chicago/Turabian StyleCasamassimi, Amelia, Antonio Federico, Monica Rienzo, Sabrina Esposito, and Alfredo Ciccodicola. 2017. "Transcriptome Profiling in Human Diseases: New Advances and Perspectives" International Journal of Molecular Sciences 18, no. 8: 1652. https://doi.org/10.3390/ijms18081652
APA StyleCasamassimi, A., Federico, A., Rienzo, M., Esposito, S., & Ciccodicola, A. (2017). Transcriptome Profiling in Human Diseases: New Advances and Perspectives. International Journal of Molecular Sciences, 18(8), 1652. https://doi.org/10.3390/ijms18081652