Increased Expression of Plasma-Induced ABCC1 mRNA in Cystic Fibrosis


Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristics of Subjects
2.2. Association between Disease Severity Categories and Clinical Status
2.3. Frequency Distribution of ABCC1 SNP rs504348 and Association with Clinical Status
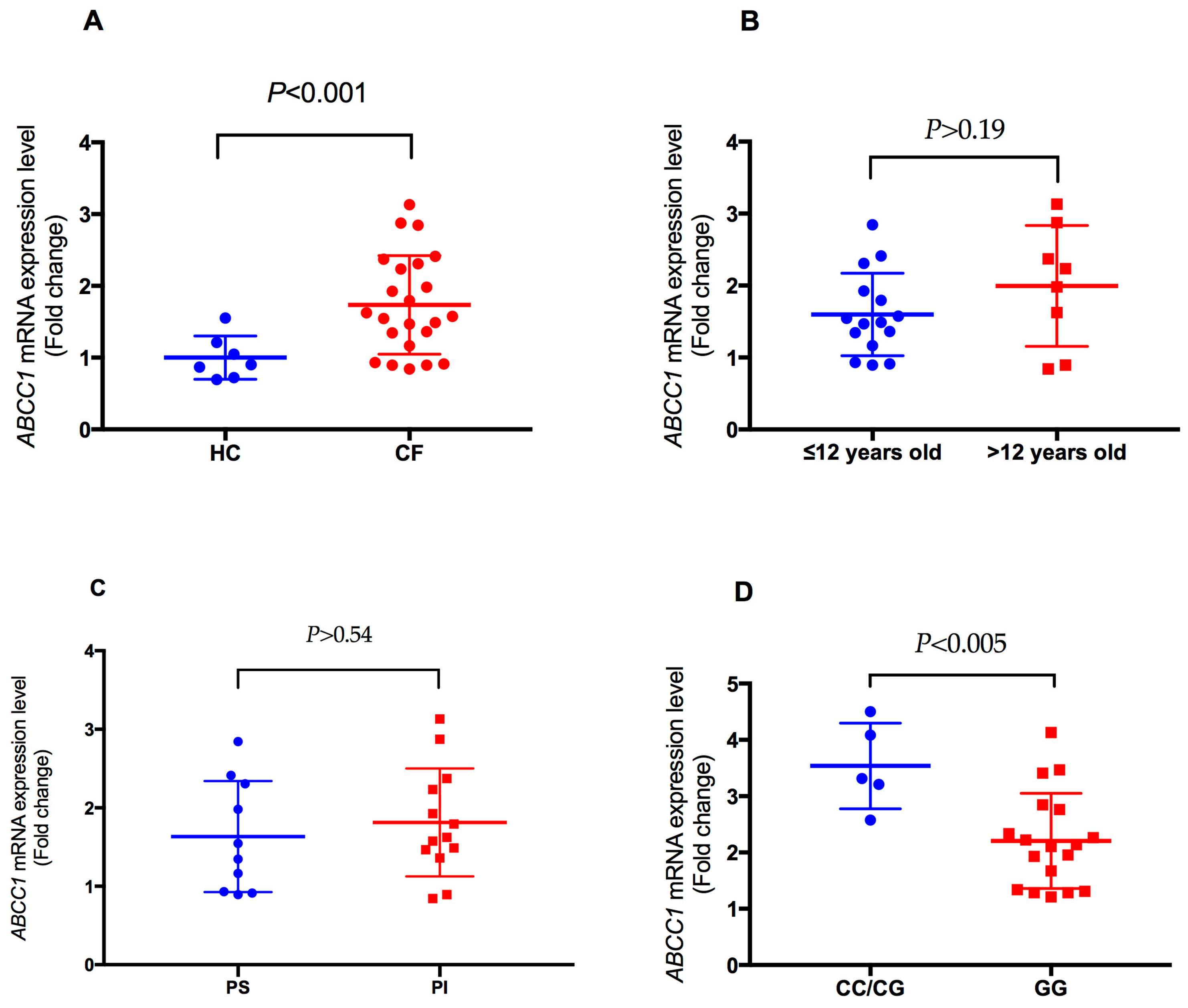
2.4. Analysis of Plasma-Induced ABCC1 mRNA Expression in CF
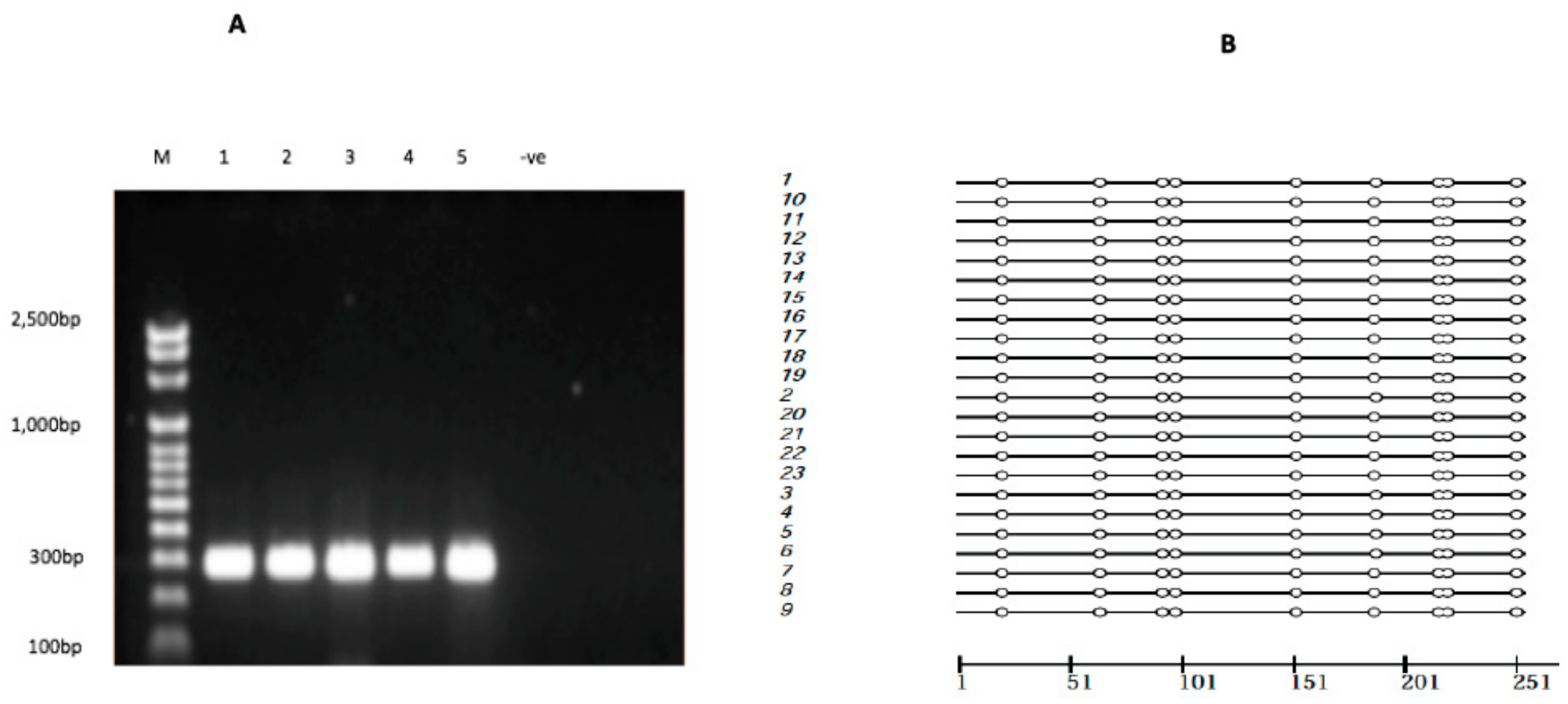
2.5. ABCC1 Promoter Methylation
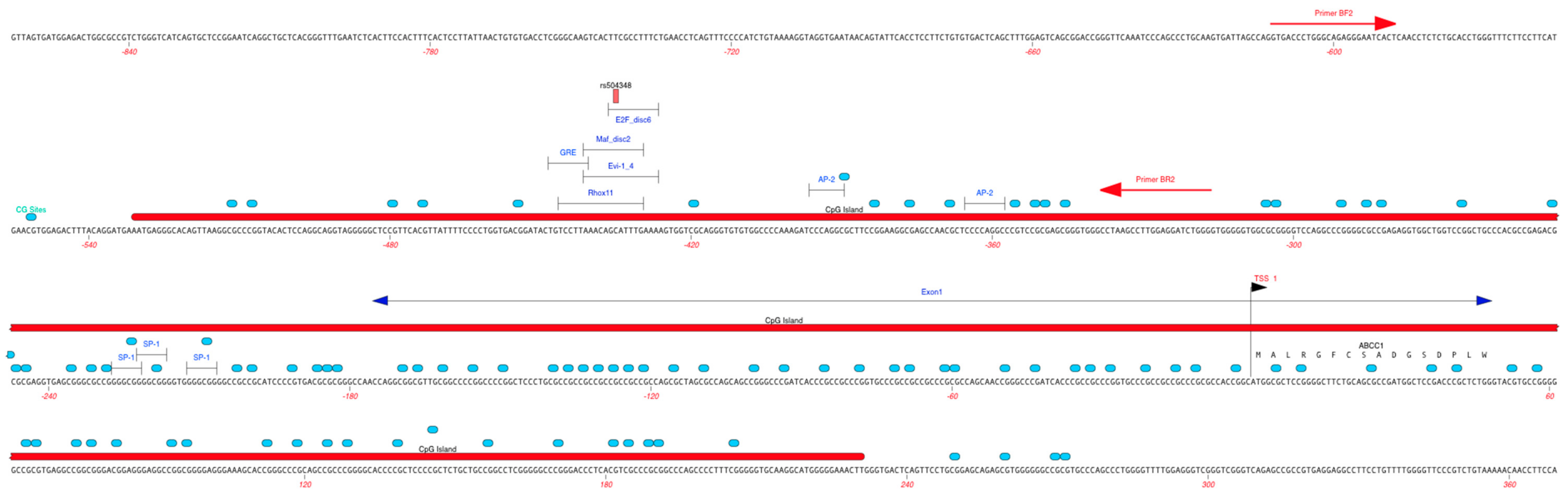
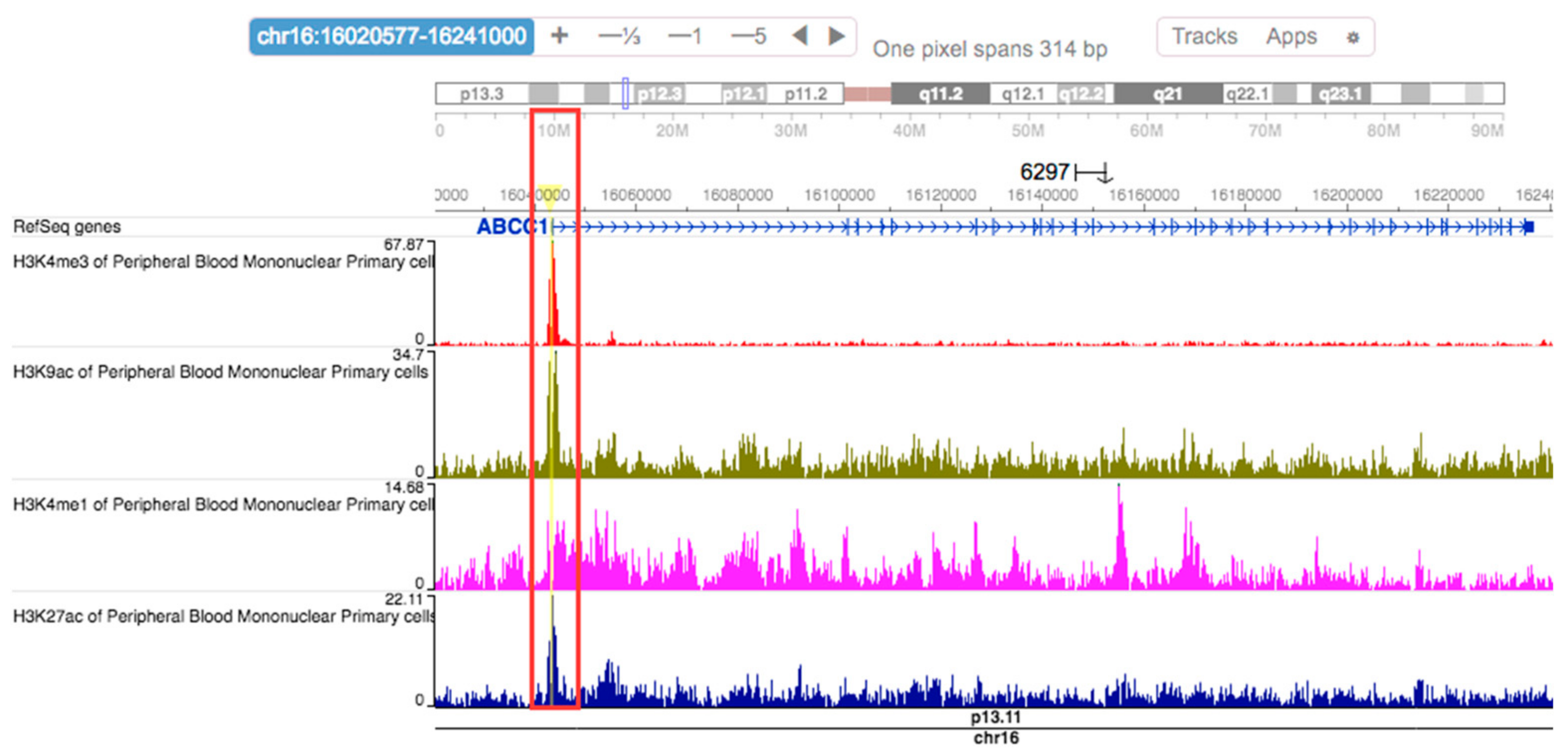
2.6. Regulatory Effects of rs504348
3. Discussion
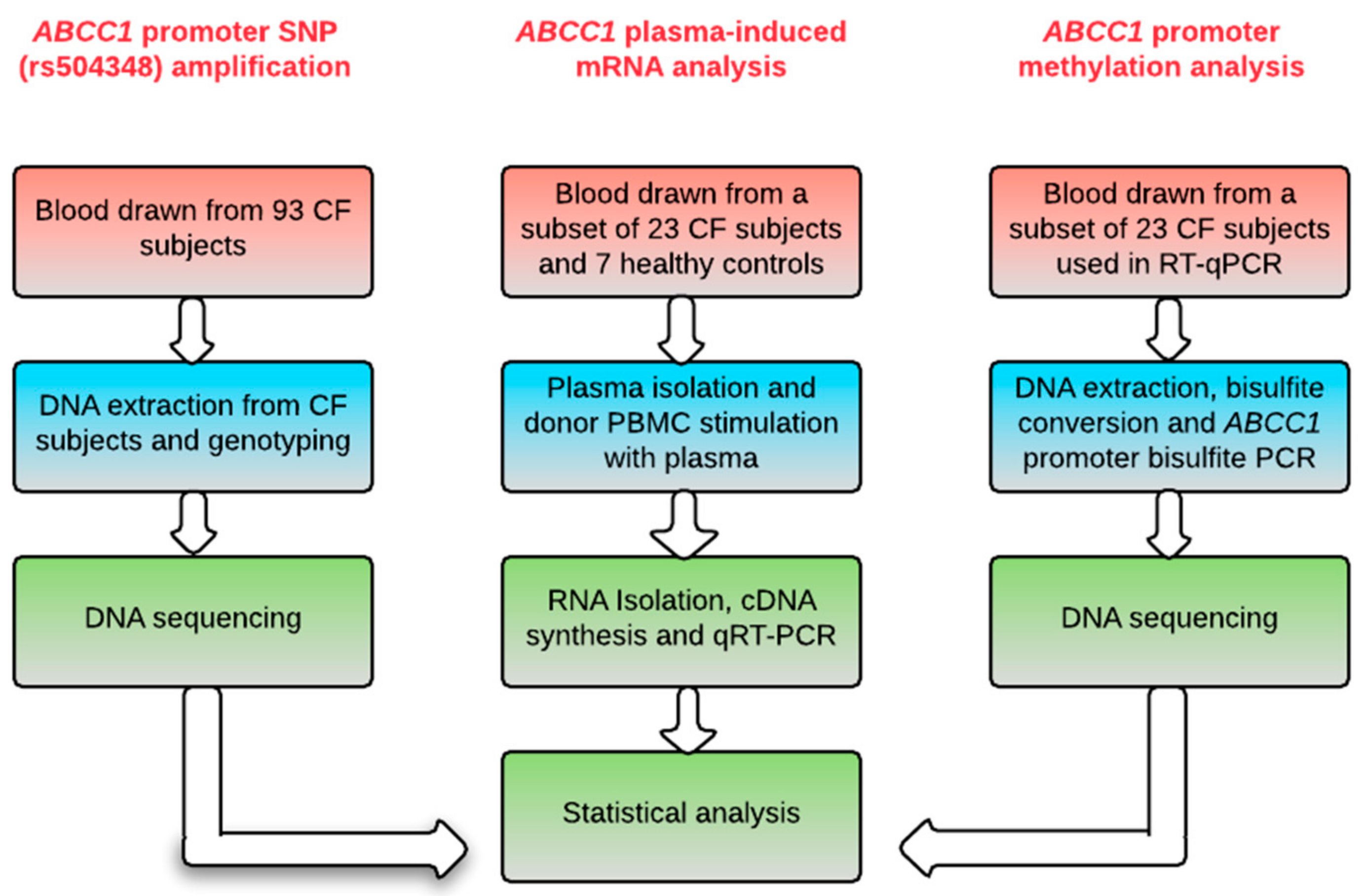
4. Materials and Methods
4.1. Study Sample Characteristics
4.2. Genotyping
4.3. PBMC Culture, Total RNA Isolation, and qRT-PCRRT-qPCR
4.4. Bisulfite PCR
4.5. SNP Annotation Data Query
4.6. DNA Sequencing and Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Gu, H.; Qiu, Y.; Guo, Y.; Korteweg, C.; Huang, J.; Gu, J. Expression of cystic fibrosis transmembrane conductance regulator in ganglia of human gastrointestinal tract. Sci. Rep. 2016, 6, 30926. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Farrell, P.M. New challenges in the diagnosis and management of cystic fibrosis. J. Pediatr. 2015, 166, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Blackman, S.M.; Boelle, P.Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015, 6, 8382. [Google Scholar] [CrossRef] [PubMed]
- Drumm, M.L.; Konstan, M.W.; Schluchter, M.D.; Handler, A.; Pace, R.; Zou, F.; Zariwala, M.; Fargo, D.; Xu, A.; Dunn, J.M.; et al. Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med. 2005, 353, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Yung, R.; Stefanowicz, D.; Shumansky, K.; Akhabir, L.; Durie, P.R.; Corey, M.; Zielenski, J.; Dorfman, R.; Daley, D.; et al. Cystic fibrosis modifier genes related to pseudomonas aeruginosa infection. Genes Immun. 2011, 12, 370–377. [Google Scholar] [CrossRef] [PubMed]
- McKone, E.F.; Shao, J.; Frangolias, D.D.; Keener, C.L.; Shephard, C.A.; Farin, F.M.; Tonelli, M.R.; Pare, P.D.; Sandford, A.J.; Aitken, M.L.; et al. Variants in the glutamate-cysteine-ligase gene are associated with cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2006, 174, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Hurbain, I.; Sermet-Gaudelus, I.; Vallee, B.; Feuillet, M.N.; Lenoir, G.; Bernaudin, J.F.; Edelman, A.; Fajac, A. Evaluation of MRP 1–5 gene expression in cystic fibrosis patients homozygous for the delta f508 mutation. Pediatr. Res. 2003, 54, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular structure of the human CFTR ion channel. Cell 2017, 169, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Schwiebert, E.M.; Benos, D.J.; Egan, M.E.; Stutts, M.J.; Guggino, W.B. CFTR is a conductance regulator as well as a chloride channel. Physiol. Rev. 1999, 79, S145–S166. [Google Scholar] [PubMed]
- Akabas, M.H. Cystic fibrosis transmembrane conductance regulator. Structure and function of an epithelial chloride channel. J. Biol. Chem. 2000, 275, 3729–3732. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.C.; Kirk, K.L. The CFTR ion channel: Gating, regulation, and anion permeation. Cold Spring Harb. Perspect. Med. 2013, 3, a009498. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, J.Y.; Stoven, V.; Annereau, J.P.; Boucher, J.; Blanquet, S.; Barthe, J.; Lenoir, G. Induction by antitumoral drugs of proteins that functionally complement CFTR: A novel therapy for cystic fibrosis? Lancet 1997, 350, 711–712. [Google Scholar] [CrossRef]
- Linsdell, P.; Hanrahan, J.W. Substrates of multidrug resistance-associated proteins block the cystic fibrosis transmembrane conductance regulator chloride channel. Br. J. Pharmacol. 1999, 126, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Center, M.S. Cloning and sequence analysis of the promoter region of the MRP gene of HL60 cells isolated for resistance to Adriamycin. Cancer Res. 1994, 54, 4488–4492. [Google Scholar] [PubMed]
- Cole, S.P.; Deeley, R.G. Multidrug resistance-associated protein: Sequence correction. Science 1993, 260, 879. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.-L.; Zhang, M.; Wang, K.; Qiao, X.-F.; Chen, M.-H. Molecular cloning, expression pattern of multidrug resistance associated protein 1 (MRP1, ABCC1) gene, and the synergistic effects of verapamil on toxicity of two insecticides in the bird cherry-oat aphid. Arch. Insect Biochem. Physiol. 2016, 92, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Pradal, U.; Delmarco, A.; Morganti, M.; Cipolli, M.; Mini, E.; Cazzola, G. Long-term azithromycin in cystic fibrosis: Another possible mechanism of action? J. Chemother. 2005, 17, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Siedlinski, M.; Boezen, H.M.; Boer, J.M.; Smit, H.A.; Postma, D.S. ABCC1 polymorphisms contribute to level and decline of lung function in two population-based cohorts. Pharmacogen. Genom. 2009, 19, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, B.; Tang, K.; Lee, E.J.; Chong, S.S.; Lee, C.G. A functional polymorphism within the MRP1 gene locus identified through its genomic signature of positive selection. Hum. Mol. Genet. 2005, 14, 2075–2087. [Google Scholar] [CrossRef] [PubMed]
- Mafficini, A.; Ortombina, M.; Sermet-Gaudelius, I.; Lebecque, P.; Leal, T.; Iansa, P.; Reychler, G.; Dahan, K.; Pepermans, X.; Lenoir, G.; et al. Impact of polymorphism of multidrug resistance-associated protein 1 (ABCC1) gene on the severity of cystic fibrosis. J. Cyst. Fibros. 2011, 10, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in hapmap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. Cpg islands—‘A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xue, X.; Wang, F.; An, Y.; Tang, D.; Xu, Y.; Wang, H.; Yuan, Z.; Gao, W.; Wei, J.; et al. Expression and promoter methylation analysis of ATP-binding cassette genes in pancreatic cancer. Oncol. Rep. 2012, 27, 265–269. [Google Scholar] [PubMed]
- Showe, M.K.; Vachani, A.; Kossenkov, A.V.; Yousef, M.; Nichols, C.; Nikonova, E.V.; Chang, C.; Kucharczuk, J.; Tran, B.; Wakeam, E.; et al. Gene expression profiles in peripheral blood mononuclear cells can distinguish patients with non-small cell lung cancer from patients with nonmalignant lung disease. Cancer Res. 2009, 69, 9202–9210. [Google Scholar] [CrossRef] [PubMed]
- Bahr, T.M.; Hughes, G.J.; Armstrong, M.; Reisdorph, R.; Coldren, C.D.; Edwards, M.G.; Schnell, C.; Kedl, R.; LaFlamme, D.J.; Reisdorph, N. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Wang, X.; Kaldunski, M.; Jia, S.; Kramer, J.; Pavletich, S.J.; Reske, M.; Gessel, T.; Yassai, M.; Quasney, M.W. Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes. Genes Immun. 2012, 13, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (Il-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to il-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Kaldunski, M.; Jia, S.; Geoffrey, R.; Basken, J.; Prosser, S.; Kansra, S.; Mordes, J.P.; Lernmark, Å.; Wang, X.; Hessner, M.J. Identification of a serum-induced transcriptional signature associated with type 1 diabetes in the biobreeding rat. Diabetes 2010, 59, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Gurram, B.; Salzman, N.H.; Kaldunski, M.L.; Jia, S.; Li, B.U.; Stephens, M.; Sood, M.R.; Hessner, M.J. Plasma-induced signatures reveal an extracellular milieu possessing an immunoregulatory bias in treatment-naive paediatric inflammatory bowel disease. Clin. Exp. Immunol. 2016, 184, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Grunau, C.; Schattevoy, R.; Mache, N.; Rosenthal, A. Methtools—A toolbox to visualize and analyze DNA methylation data. Nucleic Acids Res. 2000, 28, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. Haploreg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eqtls as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, W.J.; Butler, S.M.; Johnson, C.A.; Colin, A.A.; FitzSimmons, S.C.; Geller, D.E.; Konstan, M.W.; Light, M.J.; Rabin, H.R.; Regelmann, W.E. Epidemiologic study of cystic fibrosis: Design and implementation of a prospective, multicenter, observational study of patients with cystic fibrosis in the us and canada. Pediatr. Pulmonol. 1999, 28, 231–241. [Google Scholar] [CrossRef]
- Levy, H.; Murphy, A.; Zou, F.; Gerard, C.; Klanderman, B.; Schuemann, B.; Lazarus, R.; Garcia, K.C.; Celedon, J.C.; Drumm, M.; et al. Il1b polymorphisms modulate cystic fibrosis lung disease. Pediatr. Pulmonol. 2009, 44, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.P. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- Cardon, L.R.; Palmer, L.J. Population stratification and spurious allelic association. Lancet 2003, 361, 598–604. [Google Scholar] [CrossRef]
- Gomes, K.F.B.; Santos, A.S.; Semzezem, C.; Correia, M.R.; Brito, L.A.; Ruiz, M.O.; Fukui, R.T.; Matioli, S.R.; Passos-Bueno, M.R.; Da Silva, M.E.R. The influence of population stratification on genetic markers associated with type 1 diabetes. Sci. Rep. 2017, 7, 43513. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.L.; Burg, T.M.; Steeves, T.E. Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PLoS ONE 2012, 7, e45170. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Corey, M.; Kerem, B.S.; Rommens, J.; Markiewicz, D.; Levison, H.; Tsui, L.C.; Durie, P. The relation between genotype and phenotype in cystic fibrosis—Analysis of the most common mutation (delta F508). N. Engl. J. Med. 1990, 323, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Dorfman, R.; Cipolli, M.; Gonska, T.; Castellani, C.; Keenan, K.; Freedman, S.D.; Zielenski, J.; Berthiaume, Y.; Corey, M. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 2011, 140, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Corey, M.; Forstner, G.; Zielenski, J.; Tsui, L.; Ellis, L.; Tullis, E.; Durie, P. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut 2003, 52, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkowiak, J.; Herzig, K.H.; Witt, M.; Pogorzelski, A.; Piotrowski, R.; Barra, E.; Sobczynska-Tomaszewska, A.; Trawinska-Bartnicka, M.; Strzykala, K.; Cichy, W. Analysis of exocrine pancreatic function in cystic fibrosis: One mild CFTR mutation does not exclude pancreatic insufficiency. Eur. J. Clin. Investig. 2001, 31, 796–801. [Google Scholar] [CrossRef]
- Cleveland, R.H.; Zurakowski, D.; Slattery, D.; Colin, A.A. Cystic fibrosis genotype and assessing rates of decline in pulmonary status. Radiology 2009, 253, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, D.B.; Wilson, K.; Ferkol, T.; Schuster, D.P. Lung function decline in cystic fibrosis patients and timing for lung transplantation referral. Chest 2004, 126, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Schluchter, M.D.; Konstan, M.W.; Drumm, M.L.; Yankaskas, J.R.; Knowles, M.R. Classifying severity of cystic fibrosis lung disease using longitudinal pulmonary function data. Am. J. Respir. Crit. Care Med. 2006, 174, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Robinson, D.; Whitehead, M.; Diderichsen, F.; Olesen, H.V.; Pressler, T.; Smyth, R.L.; Diggle, P. Understanding the natural progression in % FEV1 decline in patients with cystic fibrosis: A longitudinal study. Thorax 2012, 67, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Com, G.; Carroll, J.L.; Castro, M.M.; Tang, X.; Jambhekar, S.; Berlinski, A. Predictors and outcome of low initial forced expiratory volume in 1 second measurement in children with cystic fibrosis. J. Pediatr. 2014, 164, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, H.G.; Benedetti, A.; Landry, J.S.; Bernier, J.; Matouk, E.; Radzioch, D.; Lands, L.C.; Rousseau, S.; Nguyen, D. Clinical outcomes associated with staphylococcus aureus and pseudomonas aeruginosa airway infections in adult cystic fibrosis patients. BMC Pulm. Med. 2015, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Jarad, N.A.; Higgs, S.; Jeffcote, T.; Giles, K. Factors associated with reduced FEV1 in adult patients with cystic fibrosis in a relatively affluent area. Chronic Respir. Dis. 2005, 2, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Słomka, M.; Sobalska-Kwapis, M.; Korycka-Machała, M.; Bartosz, G.; Dziadek, J.; Strapagiel, D. Genetic variation of the ABC transporter gene ABCC1 (multidrug resistance protein 1–MRP1) in the Polish population. BMC Genet. 2015, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. Cpg-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.; Francis, J.; Rosenthal, M.; Bush, A. Long-term azithromycin may improve lung function in children with cystic fibrosis. Lancet 1998, 351, 420. [Google Scholar] [CrossRef]
- Saiman, L.; Marshall, B.C.; Mayer-Hamblett, N.; Burns, J.L.; Quittner, A.L.; Cibene, D.A.; Coquillette, S.; Fieberg, A.Y.; Accurso, F.J.; Campbell, P.W., III. Azithromycin in patients with cystic fibrosis chronically infected with pseudomonas aeruginosa: A randomized controlled trial. JAMA 2003, 290, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chai, D.; Wang, R.; Bai, N.; Liang, B.B.; Liu, Y. Effectiveness and safety of macrolides in cystic fibrosis patients: A meta-analysis and systematic review. J. Antimicrob. Chemother. 2011, 66, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, E.L. Azithromycin, the multidrug-resistant protein, and cystic fibrosis. Lancet 1998, 351, 1286. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. The multidrug resistance protein family. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1461, 347–357. [Google Scholar] [CrossRef]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 690–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strange, R.C.; Spiteri, M.A.; Ramachandran, S.; Fryer, A.A. Glutathione-S-transferase family of enzymes. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2001, 482, 21–26. [Google Scholar] [CrossRef]
- Baranov, V.; Ivaschenko, T.; Bakay, B.; Aseev, M.; Belotserkovskaya, R.; Baranova, H.; Malet, P.; Perriot, J.; Mouraire, P.; Baskakov, V. Proportion of the GSTM 10/0 genotype in some slavic populations and its correlation with cystic fibrosis and some multifactorial diseases. Hum. Genet. 1996, 97, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzo, C.S.; Ribeiro, A.F.; Ribeiro, J.D. Polymorphisms in the glutathione pathway modulate cystic fibrosis severity: A cross-sectional study. BMC Med. Genet. 2014, 15, 27. [Google Scholar]
- Davies, J.C. Pseudomonas aeruginosa in cystic fibrosis: Pathogenesis and persistence. Paediatr. Respir. Rev. 2002, 3, 128–134. [Google Scholar] [CrossRef]
- Van Ewijk, B.; Wolfs, T.; Fleer, A.; Kimpen, J.; Van der Ent, C. High pseudomonas aeruginosa acquisition rate in cf. Thorax 2006, 61, 641–642. [Google Scholar] [CrossRef] [PubMed]
- Yum, H.-K.; Park, I.-N.; Shin, B.-M.; Choi, S.-J. Recurrent pseudomonas aeruginosa infection in chronic lung diseases: Relapse or reinfection? Tuberc. Respir. Dis. 2014, 77, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Conway, S.P.; Brownlee, K.G.; Denton, M.; Peckham, D.G. Antibiotic treatment of multidrug-resistant organisms in cystic fibrosis. Am. J. Respir. Med. 2003, 2, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Ang, J.Y.; Abdel-Haq, N.; Zhu, F.; Thabit, A.K.; Nicolau, D.P.; Satlin, M.J.; van Duin, D. Multidrug-resistant pseudomonas aeruginosa infection in a child with cystic fibrosis. Antimicrob. Agents Chemother. 2016, 60, 5627–5630. [Google Scholar] [CrossRef] [PubMed]
- Cândido, P.H.C.; de Souza Nunes, L.; Marques, E.A.; Folescu, T.W.; Coelho, F.S.; de Moura, V.C.N.; da Silva, M.G.; Gomes, K.M.; da Silva Lourenço, M.C.; Aguiar, F.S. Multidrug-resistant nontuberculous mycobacteria isolated from cystic fibrosis patients. J. Clin. Microbiol. 2014, 52, 2990–2997. [Google Scholar] [CrossRef] [PubMed]
- Van Dorn, A. Multidrug-Resistant Mycobacterium Abscessus Threatens Patients with Cystic Fibrosis; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Bryant, J.M.; Grogono, D.M.; Rodriguez-Rincon, D.; Everall, I.; Brown, K.P.; Moreno, P.; Verma, D.; Hill, E.; Drijkoningen, J.; Gilligan, P. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 2016, 354, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Ideozu, E.J.; Whiteoak, A.M.; Tomlinson, A.J.; Robertson, A.; Delahay, R.J.; Hide, G. High prevalence of trypanosomes in European badgers detected using its-PCR. Parasites Vectors 2015, 8, 480. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(t)(-delta delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-C.; Dahiya, R. Methprimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Maricque, B.; Xie, M.C.; Li, D.F.; Sundaram, V.; Martin, E.A.; Koebbe, B.C.; Nielsen, C.; Hirst, M.; Farnham, P.; et al. The human epigenome browser at Washington university. Nat. Methods 2011, 8, 989–990. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CF Subjects |
|---|---|
| Samples (n) | 93 |
| Age in years, median (IQR) | 10 (6,19) |
| Gender: Male, n (%) | 44 (47.3%) |
| Pancreatic insufficient, n (%) | 69 (74.2%) |
| P. aeruginosa, n (%) | 54 (58.1%) |
| Mucoid P. aeruginosa, n (%) | 32 (34.4%) |
| Sweat chloride (mmol/L), median (IQR) | 103 (87,114) |
| FEV1 (%) predicted, median (IQR) | 97 (72,111) |
| ESCF disease severity classification | n (%) |
| Normal/very mild | 44 (47.3%) |
| Mild | 10 (10.8%) |
| Moderate | 7 (7.5%) |
| Severe | 19 (20.4%) |
| Uncharacterized 1 | 13 (14.0%) |
| CFTR genotypes | n (%) |
| F508del homozygotes | 45 (48.4%) |
| F508del heterozygotes | 40 (43.0%) |
| Other 2 | 8 (8.6%) |
| Parameter | Status | ESCF Disease Severity Classification n (%) | Pancreatic Status n (%) | ||||
|---|---|---|---|---|---|---|---|
| Severe | Moderate | Mild | Normal/Very Mild | PI | PS | ||
| P. aeruginosa status *,§ | Positive | 15 (79) | 7 (100) | 5 (50) | 23 (52) | 45 (65) | 9 (38) |
| Mucoid P. aeruginosa status *,§ | Positive | 12 (63) | 5 (71) | 3 (30) | 12 (27) | 29 (42) | 3 (13) |
| Pancreatic status * | Pancreatic insufficient | 19 (100) | 6 (86) | 10 (100) | 28 (64) | -- | -- |
| Parameter | Genotype | HWE p-Value | p-Value3 | ||
|---|---|---|---|---|---|
| CC | CG | GG | |||
| Populations 1 | n (%) | ||||
| Global population | 360 (14.4%) | 658 (26.3%) | 1486 (59.3%) | <0.0001 | -- |
| CEU | 6 (6.1%) | 22 (22.2%) | 71 (71.7%) | <0.03 | -- |
| Study cohort | 8 (8.6%) | 24 (25.8%) | 61 (65.6%) | <0.03 | -- |
| ESCF Disease Severity Classification | n (%) | >0.17 | |||
| Normal/very mild | 5 (5.4%) | 9 (9.7%) | 30 (32.3%) | -- | |
| Mild | 1 (1.1%) | 3 (3.2%) | 6 (6.5%) | -- | |
| Moderate | 0 (0%) | 1 (1.1%) | 6 (6.5%) | -- | |
| Severe | 2 (2.2%) | 3 (3.2%) | 14 (15.1%) | -- | |
| Uncharacterized 2 | 0 (0%) | 8 (8.6%) | 5 (5.4%) | -- | |
| Pancreatic Status | n (%) | >0.14 | |||
| Pancreatic insufficient | 5 (5.4%) | 15 (16.1%) | 49 (52.7%) | -- | |
| Pancreatic sufficient | 3 (3.2%) | 9 (9.7%) | 12 (12.9%) | -- | |
| P. aeruginosa | n (%) | >0.89 | |||
| Positive | 4 (4.3%) | 14 (15.1%) | 36 (38.7%) | -- | |
| Negative | 4 (4.3%) | 10 (10.8%) | 25 (26.9%) | -- | |
| Mucoid P. aeruginosa | n (%) | >0.31 | |||
| Positive | 4 (4.3%) | 10 (10.8%) | 18 (19.4%) | -- | |
| Negative | 4 (4.3%) | 14 (15.1%) | 43 (46.2%) | -- | |
| Parameter | CF Subjects | Healthy Controls 1 |
|---|---|---|
| Samples, n | 23 | 7 |
| Age in years, median (IQR) | 9.4 (6.7, 17.3) | 26.9 (7.0, 27.1) |
| Pancreatic insufficient, n (%) | 13 (56.5%) | -- |
| ABCC1 genotypes | n (%) | |
| Ancestral GG | 5 (21.7%) | -- |
| rs504348 (CC/CG) | 18 (78.3%) | -- |
| CFTR genotypes 2 | n (%) | |
| F508del homozygotes | 11 (48.0%) | -- |
| F508del heterozygotes | 11 (48.0%) | -- |
| G542X/W1282X | 1 (4.0%) | -- |
| hg38 Chromosome (Position) | SNP | SNPs High LD (r2) | Promoter Histone Marks | Enhancer Histone Marks | DNAse | Proteins Bound | Motifs Changed | eQTL Hits |
|---|---|---|---|---|---|---|---|---|
| 16 (15949317) | rs504348 | 1 | 24 tissues, including PBMCs | Many tissues including PBMCs | 30 tissues | 8 bound proteins | 4 altered motifs | 2 hits 2 |
| 16 (15954300) | rs762775 1 | 0.62 | 13 tissues | 1 tissue | 2 altered motifs | 2 hits |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ideozu, J.E.; Zhang, X.; Pan, A.; Ashrafi, Z.; Woods, K.J.; Hessner, M.J.; Simpson, P.; Levy, H. Increased Expression of Plasma-Induced ABCC1 mRNA in Cystic Fibrosis. Int. J. Mol. Sci. 2017, 18, 1752. https://doi.org/10.3390/ijms18081752
Ideozu JE, Zhang X, Pan A, Ashrafi Z, Woods KJ, Hessner MJ, Simpson P, Levy H. Increased Expression of Plasma-Induced ABCC1 mRNA in Cystic Fibrosis. International Journal of Molecular Sciences. 2017; 18(8):1752. https://doi.org/10.3390/ijms18081752
Chicago/Turabian StyleIdeozu, Justin E., Xi Zhang, Amy Pan, Zainub Ashrafi, Katherine J. Woods, Martin J. Hessner, Pippa Simpson, and Hara Levy. 2017. "Increased Expression of Plasma-Induced ABCC1 mRNA in Cystic Fibrosis" International Journal of Molecular Sciences 18, no. 8: 1752. https://doi.org/10.3390/ijms18081752
APA StyleIdeozu, J. E., Zhang, X., Pan, A., Ashrafi, Z., Woods, K. J., Hessner, M. J., Simpson, P., & Levy, H. (2017). Increased Expression of Plasma-Induced ABCC1 mRNA in Cystic Fibrosis. International Journal of Molecular Sciences, 18(8), 1752. https://doi.org/10.3390/ijms18081752