Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids


Abstract
:1. Introduction
2. Mechanisms of GC Action
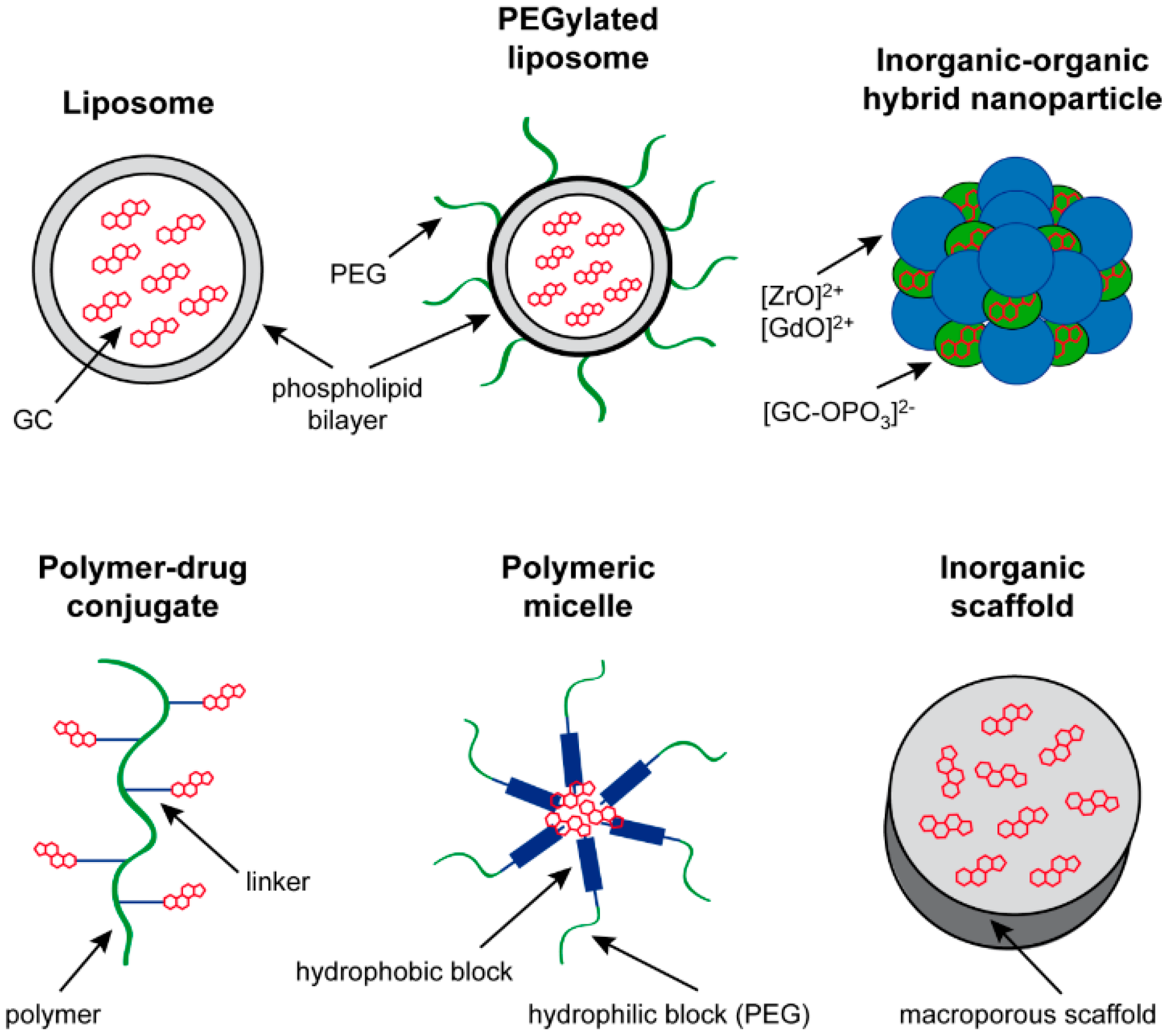
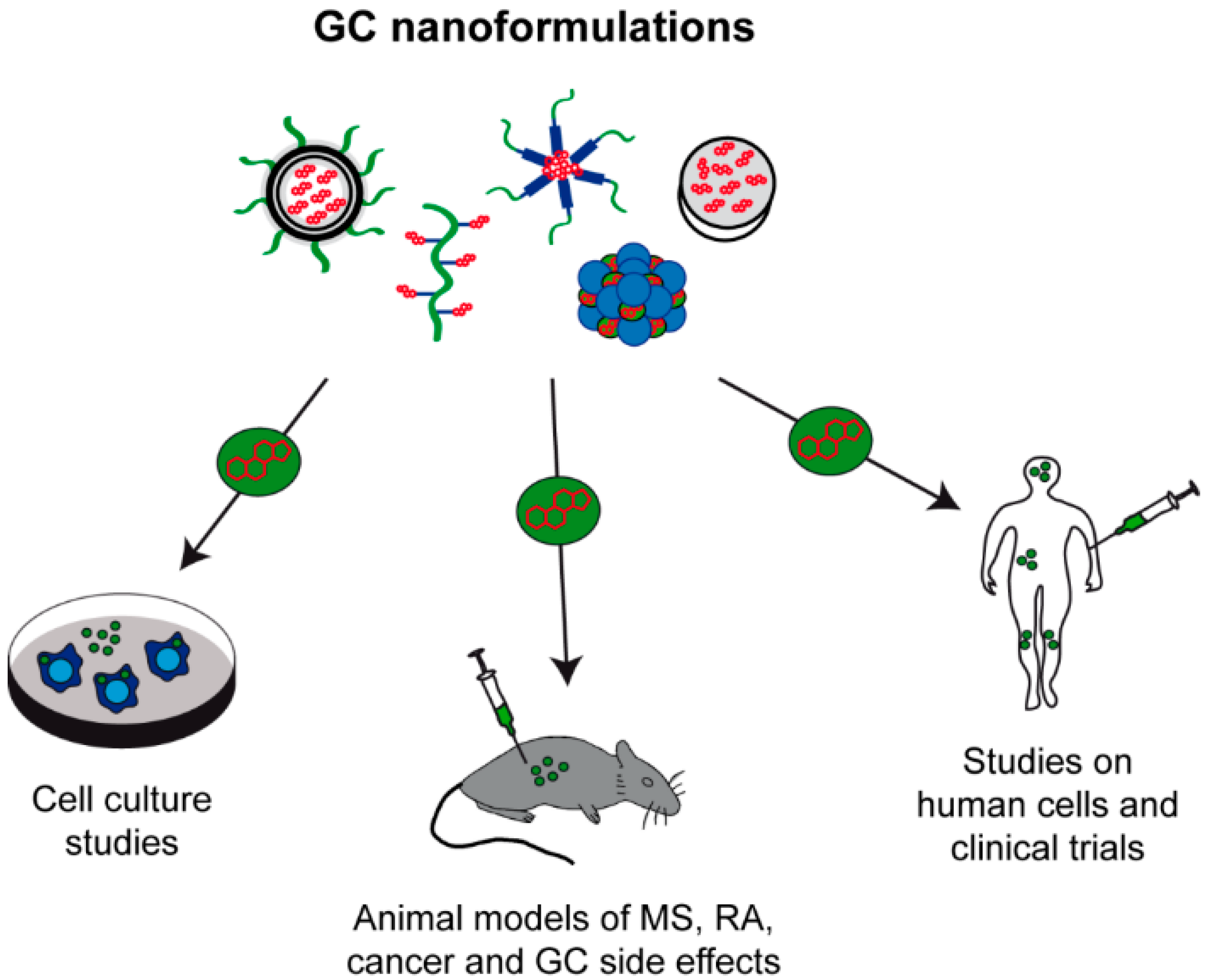
3. Drug Delivery Systems for GC Application
3.1. Liposomes
3.2. Polymeric Micelles
3.3. Polymer-Drug Conjugates
3.4. Inorganic Drug Delivery Systems
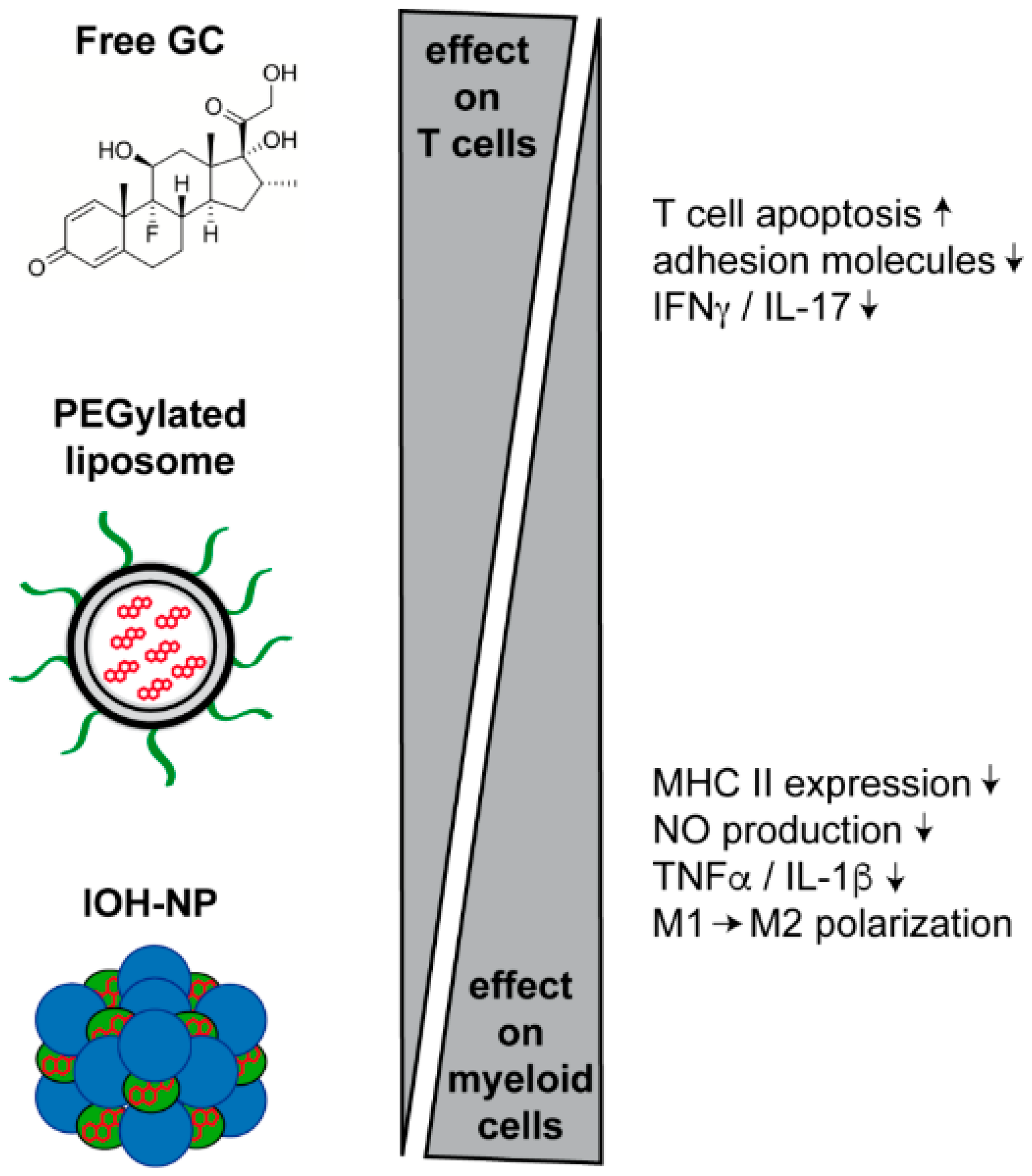
3.5. Inorganic–Organic NP
4. GC Nanoformulations in the Treatment of Disorders of the CNS
4.1. Treatment of Neuroinflammatory Diseases
4.2. Treatment of Other CNS Diseases
5. Alternative GC Delivery in the Treatment of Other Diseases
5.1. Cancer
5.2. Rheumatoid Arthritis
5.3. Application of GC Nanoformulations in Other Preclinical Studies
6. From Bench to Bedside-Transfer to the Clinic
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BMP | Betamethasone phosphate |
| Bud | Budenoside |
| CNS | Central nervous system |
| Dex | Dexamethasone |
| EAE | Experimental autoimmune encephalomyelitis |
| EPR | Enhanced permeability and retention effect |
| HPMA | N-(2-hydroxypropyl) methacrylamide |
| GC | Glucocorticoids |
| GR | Glucocorticoid receptor |
| GRE | Glucocorticoid response element |
| IL | Interleukin |
| IOH-NP | Inorganic-organic hybrid nanoparticles |
| LPS | Lipopolysaccharide |
| MP | Methylprednisolone |
| MRI | Magnetic resonance imaging |
| MRT | Magnetic resonance tomography |
| MS | Multiple sclerosis |
| NP | Nanoparticles |
| PDN | Prednisolone |
| PEG | Polyethylene glycol |
| PCL | Poly ε-caprolactone |
| RA | Rheumatoid arthritis |
| TAM | Tumor-associated macrophages |
| TNF | Tumor necrosis factor |
References
- Hench, P.S.; Kendall, E.C.; Slocumb, C.H.; Polley, H.F. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: Compound-E) and of pituitary adrenocorticotrophic hormone on rheumatoid arthritis-preliminary report. Ann. Rheum. Dis. 1949, 8, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hench, P.S.; Kendall, E.C. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: Compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc. Staff. Meet. Mayo Clin. 1949, 24, 181–197. [Google Scholar] [PubMed]
- Hao, H.-X.; Wang, J.-K.; Wang, J.-L. Solubility of dexamethasone sodium phosphate in different solvents. J. Chem. Eng. Data 2004, 49, 1697–1698. [Google Scholar] [CrossRef]
- Granner, D.K.; Wang, J.C.; Yamamoto, K.R. Regulatory actions of glucocorticoid hormones: From organisms to mechanisms. Adv. Exp. Med. Biol. 2015, 872, 3–31. [Google Scholar] [PubMed]
- Buttgereit, F.; Spies, C.M.; Bijlsma, J.W. Novel glucocorticoids: Where are we now and where do we want to go? Clin. Exp. Rheumatol. 2015, 33, S29–S33. [Google Scholar] [PubMed]
- Rauch, A.; Seitz, S.; Baschant, U.; Schilling, A.F.; Illing, A.; Stride, B.; Kirilov, M.; Mandic, V.; Takacz, A.; Schmidt-Ullrich, R.; et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010, 11, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.; Fett, N.; Rosenbach, M.; Werth, V.P.; Micheletti, R.G. Prevention and management of glucocorticoid-induced side effects: A comprehensive review: Ocular, cardiovascular, muscular, and psychiatric side effects and issues unique to pediatric patients. J. Am. Acad. Dermatol. 2017, 76, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.D.; Weinhage, T.; Rotte, A.; Foller, M.; Oppermann, M.; Lühder, F.; Tuckermann, J.P.; Lang, F.; van den Brandt, J.; Reichardt, H.M. Glucocorticoids induce gastroparesis in mice through depletion of l-arginine. Endocrinology 2014, 155, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.L.; Baehr, L.M.; Reichardt, H.M.; Tuckermann, J.P.; Bodine, S.C.; Furlow, J.D. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1210–E1220. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Song, I.H.; Gold, R.; Straub, R.H.; Burmester, G.R.; Buttgereit, F. New glucocorticoids on the horizon: Repress, don’t activate! J. Rheumatol. 2005, 32, 1199–1207. [Google Scholar] [PubMed]
- Schaecke, H.; Berger, M.; Rehwinkel, H.; Asadullah, K. Selective glucocorticoid receptor agonists (SEGRAs): Novel ligands with an improved therapeutic index. Mol. Cell Endocrinol. 2007, 275, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Paul-Clark, M.J.; Mancini, L.; Del, S.P.; Flower, R.J.; Perretti, M. Potent antiarthritic properties of a glucocorticoid derivative, NCX-1015, in an experimental model of arthritis. Proc. Natl. Acad. Sci. USA 2002, 99, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Avery, W.; Finelli, A.L.; Farwell, M.; Fraser, C.C.; Borisy, A.A. Selective amplification of glucocorticoid anti-inflammatory activity through synergistic multi-target action of a combination drug. Arthritis Res. Ther. 2009, 11, R12. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Doering, G.; Schaeffler, A.; Witte, S.; Sierakowski, S.; Gromnica-Ihle, E.; Jeka, S.; Krueger, K.; Szechinski, J.; Alten, R. Efficacy of modified-release versus standard prednisone to reduce duration of morning stiffness of the joints in rheumatoid arthritis (CAPRA-1): A double-blind, randomised controlled trial. Lancet 2008, 371, 205–214. [Google Scholar] [CrossRef]
- Buttgereit, F.; Doering, G.; Schaeffler, A.; Witte, S.; Sierakowski, S.; Gromnica-Ihle, E.; Jeka, S.; Krueger, K.; Szechinski, J.; Alten, R. Targeting pathophysiological rhythms: Prednisone chronotherapy shows sustained efficacy in rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Wakaskar, R.R. General overview of lipid-polymer hybrid nanoparticles, dendrimers, micelles, liposomes, spongosomes and cubosomes. J. Drug Target 2017. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Baatar, D.; Collins, G.; Carter, A.; Indig, F.; Biragyn, A.; Taub, D.D. Dexamethasone augments CXCR4-mediated signaling in resting human T cells via the activation of the Src kinase Lck. Blood 2009, 113, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Fischer, H.J.; Tischner, D.; van den Brandt, J.; Reichardt, H.M. Glucocorticoids induce effector T cell depolarization via ERM proteins, thereby impeding migration and APC conjugation. J. Immunol. 2013, 190, 4360–4370. [Google Scholar] [CrossRef] [PubMed]
- Hübner, S.; Dejager, L.; Libert, C.; Tuckermann, J.P. The glucocorticoid receptor in inflammatory processes: Transrepression is not enough. Biol. Chem. 2015, 396, 1223–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Van, B.T.; Kleyman, A.; Liu, Y.; Tuckermann, J.; Libert, C. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J. Clin. Investig. 2012, 122, 2130–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [PubMed]
- Uhlenhaut, N.H.; Barish, G.D.; Yu, R.T.; Downes, M.; Karunasiri, M.; Liddle, C.; Schwalie, P.; Hübner, N.; Evans, R.M. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol. Cell 2013, 49, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Schottelius, A.; Döcke, W.D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc. Natl. Acad. Sci. USA 2004, 101, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wüst, S.; Tischner, D.; John, M.; Tuckermann, J.P.; Menzfeld, C.; Hanisch, U.K.; van den Brandt, J.; Lühder, F.; Reichardt, H.M. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS ONE 2009, 4, e8202. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; Schütz, G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef]
- Schweingruber, N.; Fischer, H.J.; Fischer, L.; van den Brandt, J.; Karabinskaya, A.; Labi, V.; Villunger, A.; Kretzschmar, B.; Huppke, P.; Simons, M.; et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014, 127, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, A.; Hübner, S.; Rodriguez Parkitna, J.M.; Neumann, A.; Hofer, S.; Weigand, M.A.; Bauer, M.; Schmid, W.; Schütz, G.; Libert, C.; et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 2012, 26, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Baschant, U.; Frappart, L.; Rauchhaus, U.; Bruns, L.; Reichardt, H.M.; Kamradt, T.; Brauer, R.; Tuckermann, J.P. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19317–19322. [Google Scholar] [CrossRef] [PubMed]
- Klaßen, C.; Karabinskaya, A.; Dejager, L.; Vettorazzi, S.; van Moorleghem, J.; Lühder, F.; Meijsing, S.H.; Tuckermann, J.P.; Bohnenberger, H.; Libert, C.; et al. Airway Epithelial cells are crucial targets of glucocorticoids in a mouse model of allergic asthma. J. Immunol. 2017, 199, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Wüst, S.; van den Brandt, J.; Tischner, D.; Kleiman, A.; Tuckermann, J.P.; Gold, R.; Lühder, F.; Reichardt, H.M. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 8434–8443. [Google Scholar] [CrossRef] [PubMed]
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.E.; Stride, B.; Förster, I.; Habenicht, A.J.R.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Investig. 2007, 117, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Triantafillou, G.; Hertlein, E.; Towns, W.; Stefanovski, M.; Mo, X.; Jarjoura, D.; Phelps, M.; Marcucci, G.; Lee, L.J.; et al. Milatuzumab-conjugated liposomes as targeted dexamethasone carriers for therapeutic delivery in CD74 + B-cell malignancies. Clin. Cancer Res. 2013, 19, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Song, Y.; Lu, M.; Lin, X.; Liu, Y.; Zhou, S.; Su, Y.; Deng, Y. Evaluation of the antitumor effect of dexamethasone palmitate and doxorubicin co-loaded liposomes modified with a sialic acid-octadecylamine conjugate. Eur. J. Pharm. Sci. 2016, 93, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Cao, H.; Yang, C.; Zhang, L.; Jiang, X.; Gao, X.; Yang, F.; He, G.; Song, X.; Tong, A.; et al. LHD-modified mechanism-based liposome coencapsulation of mitoxantrone and prednisolone using novel lipid bilayer fusion for tissue-specific colocalization and synergistic antitumor effects. ACS Appl. Mater. Interfaces 2016, 8, 6586–6601. [Google Scholar] [CrossRef] [PubMed]
- Schiffelers, R.M.; Metselaar, J.M.; Fens, M.H.; Janssen, A.P.; Molema, G.; Storm, G. Liposome-encapsulated prednisolone phosphate inhibits growth of established tumors in mice. Neoplasia 2005, 7, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Avnir, Y.; Turjeman, K.; Tulchinsky, D.; Sigal, A.; Kizelsztein, P.; Tzemach, D.; Gabizon, A.; Barenholz, Y. Fabrication principles and their contribution to the superior in vivo therapeutic efficacy of nano-liposomes remote loaded with glucocorticoids. PLoS ONE 2011, 6, e25721. [Google Scholar] [CrossRef] [PubMed]
- Kroon, J.; Buijs, J.T.; van der Horst, G.; Cheung, H.; van der Mark, M.; van, B.L.; Rizzo, L.Y.; Lammers, T.; Pelger, R.C.; Storm, G.; et al. Liposomal delivery of dexamethasone attenuates prostate cancer bone metastatic tumor growth in vivo. Prostate 2015, 75, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Banciu, M.; Metselaar, J.M.; Schiffelers, R.M.; Storm, G. Liposomal glucocorticoids as tumor-targeted anti-angiogenic nanomedicine in B16 melanoma-bearing mice. J. Steroid Biochem. Mol. Biol. 2008, 111, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Banciu, M.; Fens, M.H.; Storm, G.; Schiffelers, R.M. Antitumor activity and tumor localization of liposomal glucocorticoids in B16 melanoma-bearing mice. J. Control. Release 2008, 127, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, M.; Rijcken, C.J.; Stigter, M.; Hennink, W.E.; Storm, G.; Schiffelers, R.M. Antitumor efficacy of dexamethasone-loaded core-crosslinked polymeric micelles. J. Control. Release 2012, 163, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kluza, E.; Yeo, S.Y.; Schmid, S.; van der Schaft, D.W.; Boekhoven, R.W.; Schiffelers, R.M.; Storm, G.; Strijkers, G.J.; Nicolay, K. Anti-tumor activity of liposomal glucocorticoids: The relevance of liposome-mediated drug delivery, intratumoral localization and systemic activity. J. Control. Release 2011, 151, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Vanniasinghe, A.S.; Manolios, N.; Schibeci, S.; Lakhiani, C.; Kamali-Sarvestani, E.; Sharma, R.; Kumar, V.; Moghaddam, M.; Ali, M.; Bender, V. Targeting fibroblast-like synovial cells at sites of inflammation with peptide targeted liposomes results in inhibition of experimental arthritis. Clin. Immunol. 2014, 151, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Hofkens, W.; Storm, G.; van den Berg, W.B.; van Lent, P.L. Liposomal targeting of glucocorticoids to the inflamed synovium inhibits cartilage matrix destruction during murine antigen-induced arthritis. Int. J. Pharm. 2011, 416, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Hofkens, W.; Grevers, L.C.; Walgreen, B.; de Vries, T.J.; Leenen, P.J.; Everts, V.; Storm, G.; van den Berg, W.B.; van Lent, P.L. Intravenously delivered glucocorticoid liposomes inhibit osteoclast activity and bone erosion in murine antigen-induced arthritis. J. Control. Release 2011, 152, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Hofkens, W.; Schelbergen, R.; Storm, G.; van den Berg, W.B.; van Lent, P.L. Liposomal targeting of prednisolone phosphate to synovial lining macrophages during experimental arthritis inhibits M1 activation but does not favor M2 differentiation. PLoS ONE 2013, 8, e54016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Geest, T.; Metselaar, J.M.; Gerrits, D.; van Lent, P.L.; Storm, G.; Laverman, P.; Boerman, O.C. [(18)]F FDG PET/CT imaging to monitor the therapeutic effect of liposome-encapsulated prednisolone in experimental rheumatoid arthritis. J. Control. Release 2015, 209, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ulmansky, R.; Turjeman, K.; Baru, M.; Katzavian, G.; Harel, M.; Sigal, A.; Naparstek, Y.; Barenholz, Y. Glucocorticoids in nano-liposomes administered intravenously and subcutaneously to adjuvant arthritis rats are superior to the free drugs in suppressing arthritis and inflammatory cytokines. J. Control. Release 2012, 160, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Zhang, Y.; Crielaard, B.J.; Dusad, A.; Lele, S.M.; Rijcken, C.J.; Metselaar, J.M.; Kostkova, H.; Etrych, T.; Ulbrich, K.; et al. Nanomedicines for inflammatory arthritis: Head-to-head comparison of glucocorticoid-containing polymers, micelles, and liposomes. ACS Nano 2014, 8, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.D.; Purdue, P.E.; Liu, X.M.; Boska, M.D.; Lele, S.M.; Thiele, G.M.; Mikuls, T.R.; Dou, H.; Goldring, S.R.; Wang, D. Development of a macromolecular prodrug for the treatment of inflammatory arthritis: Mechanisms involved in arthrotropism and sustained therapeutic efficacy. Arthritis Res. Ther. 2010, 12, R170. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoven, J.M.; Hofkens, W.; Wauben, M.H.; Wagenaar-Hilbers, J.P.; Beijnen, J.H.; Nuijen, B.; Metselaar, J.M.; Storm, G. Optimizing the therapeutic index of liposomal glucocorticoids in experimental arthritis. Int. J. Pharm. 2011, 416, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Rauchhaus, U.; Kinne, R.W.; Pohlers, D.; Wiegand, S.; Wolfert, A.; Gajda, M.; Brauer, R.; Panzner, S. Targeted delivery of liposomal dexamethasone phosphate to the spleen provides a persistent therapeutic effect in rat antigen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 1933–1934. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Franch, A.; Castell, M.; Perez-Cano, F.J.; Brauer, R.; Pohlers, D.; Gajda, M.; Siskos, A.P.; Katsila, T.; Tamvakopoulos, C.; et al. Liposomal encapsulation enhances and prolongs the anti-inflammatory effects of water-soluble dexamethasone phosphate in experimental adjuvant arthritis. Arthritis Res. Ther. 2010, 12, R147. [Google Scholar] [CrossRef] [PubMed]
- Tentillier, N.; Etzerodt, A.; Olesen, M.N.; Rizalar, F.S.; Jacobsen, J.; Bender, D.; Moestrup, S.K.; Romero-Ramos, M. Anti-inflammatory modulation of microglia via CD163-targeted glucocorticoids protects dopaminergic neurons in the 6-OHDA parkinson's disease model. J. Neurosci. 2016, 36, 9375–9390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.; Metselaar, J.M.; Wauben, M.H.M.; Toyka, K.V.; Storm, G.; Gold, R. Drug targeting by long-circulating liposomal glucocorticosteroids increases therapeutic efficacy in a model of multiple sclerosis. Brain 2003, 126, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Weller, C.; Lühder, F.; Mohr, A.; Schmidt, J.; Knauth, M.; Metselaar, J.M.; Gold, R. Liposomal glucocorticosteroids in treatment of chronic autoimmune demyelination: Long-term protective effects and enhanced efficacy of methylprednisolone formulations. Exp. Neurol. 2008, 211, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Schweingruber, N.; Haine, A.; Tiede, K.; Karabinskaya, A.; van den Brandt, J.; Wüst, S.; Metselaar, J.M.; Gold, R.; Tuckermann, J.P.; Reichardt, H.M.; Lühder, F. Liposomal encapsulation of glucocorticoids alters their mode of action in the treatment of experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 4310–4318. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Rotger, C.; Appeldoorn, C.C.; Reijerkerk, A.; Gladdines, W.; Gaillard, P.J.; Linker, R.A. Glutathione PEGylated liposomal methylprednisolone (2B3–201) attenuates CNS inflammation and degeneration in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 274, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; Appeldoorn, C.C.; Rip, J.; Dorland, R.; van der Pol, S.M.; Kooij, G.; de Vries, H.E.; Reijerkerk, A. Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. J. Control. Release 2012, 164, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Turjeman, K.; Bavli, Y.; Kizelsztein, P.; Schilt, Y.; Allon, N.; Katzir, T.B.; Sasson, E.; Raviv, U.; Ovadia, H.; Barenholz, Y. Nano-Drugs Based on Nano Sterically Stabilized Liposomes for the Treatment of Inflammatory Neurodegenerative Diseases. PLoS ONE 2015, 10, e0130442. [Google Scholar] [CrossRef] [PubMed]
- Tiebosch, I.A.; Crielaard, B.J.; Bouts, M.J.; Zwartbol, R.; Salas-Perdomo, A.; Lammers, T.; Planas, A.M.; Storm, G.; Dijkhuizen, R.M. Combined treatment with recombinant tissue plasminogen activator and dexamethasone phosphate-containing liposomes improves neurological outcome and restricts lesion progression after embolic stroke in rats. J. Neurochem. 2012, 123 (Suppl. S2), 65–74. [Google Scholar] [CrossRef] [PubMed]
- Konduri, K.S.; Nandedkar, S.; Duzgunes, N.; Suzara, V.; Artwohl, J.; Bunte, R.; Gangadharam, P.R. Efficacy of liposomal budesonide in experimental asthma. J. Allergy Clin. Immunol. 2003, 111, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Konduri, K.S.; Nandedkar, S.; Rickaby, D.A.; Duzgunes, N.; Gangadharam, P.R. The use of sterically stabilized liposomes to treat asthma. Methods Enzymol. 2005, 391, 413–427. [Google Scholar] [PubMed]
- Bartneck, M.; Scheyda, K.M.; Warzecha, K.T.; Rizzo, L.Y.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, J.; Chen, W.; Jiang, H.; Zhang, Z.; Sun, X. Targeted delivery of low-dose dexamethasone using PCL-PEG micelles for effective treatment of rheumatoid arthritis. J. Control. Release 2016, 230, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Quan, L.D.; Tian, J.; Alnouti, Y.; Fu, K.; Thiele, G.M.; Wang, D. Synthesis and evaluation of a well-defined HPMA copolymer-dexamethasone conjugate for effective treatment of rheumatoid arthritis. Pharm. Res. 2008, 25, 2910–2919. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.D.; Yuan, F.; Liu, X.M.; Huang, J.G.; Alnouti, Y.; Wang, D. Pharmacokinetic and biodistribution studies of N-(2-hydroxypropyl)methacrylamide copolymer-dexamethasone conjugates in adjuvant-induced arthritis rat model. Mol. Pharm. 2010, 7, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Zhang, Y.; Dusad, A.; Ren, K.; Purdue, P.E.; Goldring, S.R.; Wang, D. The evaluation of the therapeutic efficacy and side effects of a macromolecular dexamethasone prodrug in the collagen-induced arthritis mouse model. Pharm. Res. 2016, 33, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, M.; Gu, L.; Li, X.; He, J.; Zhou, L.; Tong, A.; Shi, J.; Zhu, H.; Xu, J.; et al. Effective improvement of the neuroprotective activity after spinal cord injury by synergistic effect of glucocorticoid with biodegradable amphipathic nanomicelles. Drug Deliv. 2017, 24, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dusad, A.; Yuan, F.; Yuan, H.; Purdue, P.E.; Fehringer, E.V.; Garvin, K.L.; Goldring, S.R.; Wang, D. Macromolecular prodrug of dexamethasone prevents particle-induced peri-implant osteolysis with reduced systemic side effects. J. Control. Release 2014, 175, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; Garcia-Martin, E.; Gil, C.; Mayoral, J.A.; Pablo, L.E.; Polo, V.; Prieto, E.; Vispe, E. Laponite as carrier for controlled in vitro delivery of dexamethasone in vitreous humor models. Eur. J. Pharm. Biopharm. 2016, 108, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gianella, A.; Jarzyna, P.A.; Mani, V.; Ramachandran, S.; Calcagno, C.; Tang, J.; Kann, B.; Dijk, W.J.; Thijssen, V.L.; Griffioen, A.W.; et al. Multifunctional nanoemulsion platform for imaging guided therapy evaluated in experimental cancer. ACS Nano 2011, 5, 4422–4433. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Maleki-Dizaji, N.; Barar, J.; Barzegar-Jalali, M.; Rameshrad, M.; Adibkia, K. Methylprednisolone acetate-loaded hydroxyapatite nanoparticles as a potential drug delivery system for treatment of rheumatoid arthritis: In vitro and in vivo evaluations. Eur. J. Pharm. Sci. 2016, 91, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Montes-Cobos, E.; Ring, S.; Fischer, H.J.; Heck, J.; Strauss, J.; Schwaninger, M.; Reichardt, S.D.; Feldmann, C.; Lühder, F.; Reichardt, H.M. Targeted delivery of glucocorticoids to macrophages in a mouse model of multiple sclerosis using inorganic-organic hybrid nanoparticles. J. Control. Release 2016, 245, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Metselaar, J.M.; Mastrobattista, E.; Storm, G. Liposomes for intravenous drug targeting: Design and applications. Mini Rev. Med. Chem. 2002, 2, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Adv. Pharm. Bull. 2017, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Papahadjopoulos, D.; Gabizon, A. Liposomes designed to avoid the reticuloendothelial system. Prog. Clin. Biol. Res. 1990, 343, 85–93. [Google Scholar] [PubMed]
- Allen, T.M.; Hansen, C.; Martin, F.; Redemann, C.; Yau-Young, A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim. Biophys. Acta 1991, 1066, 29–36. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Maeda, H.; Sawa, T.; Konno, T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J. Control. Release 2001, 74, 47–61. [Google Scholar] [CrossRef]
- Everts, M.; Koning, G.A.; Kok, R.J.; Asgeirsdottir, S.A.; Vestweber, D.; Meijer, D.K.; Storm, G.; Molema, G. In vitro cellular handling and in vivo targeting of E-selectin-directed immunoconjugates and immunoliposomes used for drug delivery to inflamed endothelium. Pharm. Res. 2003, 20, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.H.; Maleki, A.; Eshraghi, H.R.; Hamidi, M. Preparation and in vitro/pharmacokinetic/pharmacodynamic evaluation of a slow-release nano-liposomal form of prednisolone. Drug Deliv. 2016, 23, 3008–3016. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Wang, X.; Shimizu, T.; Nawata, K.; Kiwada, H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J. Control. Release 2007, 122, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoven, J.M.; Nemes, R.; Metselaar, J.M.; Nuijen, B.; Beijnen, J.H.; Storm, G.; Szebeni, J. Complement activation by PEGylated liposomes containing prednisolone. Eur. J. Pharm. Sci. 2013, 49, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, A.M.; Torchilin, V.P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5, 77. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T. Improving the efficacy of combined modality anticancer therapy using HPMA copolymer-based nanomedicine formulations. Adv. Drug Deliv. Rev. 2010, 62, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Subr, V.; Ulbrich, K.; Peschke, P.; Huber, P.E.; Hennink, W.E.; Storm, G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials 2009, 30, 3466–3475. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Tong, R. Anticancer nanoparticulate polymer-drug conjugate. Bioeng. Transl. Med. 2016, 1, 277–296. [Google Scholar] [CrossRef]
- Pang, X.; Du, H.L.; Zhang, H.Q.; Zhai, Y.J.; Zhai, G.X. Polymer-drug conjugates: Present state of play and future perspectives. Drug Discov. Today 2013, 18, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Li, F.; Zhao, G.; Chhonker, Y.S.; Averill, C.; Galdamez, J.; Purdue, P.E.; Wang, X.; Fehringer, E.V.; Garvin, K.L.; et al. Pharmacokinetic and biodistribution studies of HPMA copolymer conjugates in an aseptic implant loosening mouse model. Mol. Pharm. 2017, 14, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Weaver, J.D.; Li, Y.; Chen, X.; Liang, J.; Stabler, C.L. Local release of dexamethasone from macroporous scaffolds accelerates islet transplant engraftment by promotion of anti-inflammatory M2 macrophages. Biomaterials 2017, 114, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, B.; Zaccarelli, E. A fresh look at the laponite phase diagram. Soft Matter 2011, 7, 1268–1286. [Google Scholar] [CrossRef]
- Heck, J.G.; Napp, J.; Simonato, S.; Mollmer, J.; Lange, M.; Reichardt, H.M.; Staudt, R.; Alves, F.; Feldmann, C. Multifunctional phosphate-based inorganic-organic hybrid nanoparticles. J. Am. Chem. Soc. 2015, 137, 7329–7336. [Google Scholar] [CrossRef] [PubMed]
- Milligan, N.M.; Newcombe, R.; Compston, D.A.S. A double-blind controlled trial of high-dose methylprednisolone in patients with multiple-sclerosis: 1. Clinical Effects. J. Neurol. Neurosurg. Psychiatry 1987, 50, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Gennari, A.; Salvadori, B.; Tognoni, A.; Conte, P.F. Rapid intravenous premedication with dexamethasone prevents hypersensitivity reactions to paclitaxel. Ann. Oncol. 1996, 7, 978–979. [Google Scholar] [CrossRef] [PubMed]
- Münstedt, K.; Borces, D.; Bohlmann, M.K.; Zygmunt, M.; von Georgi, R. Glucocorticoid administration in antiemetic therapy: Is it safe? Cancer 2004, 101, 1696–1702. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Luigi, M. Corticosteroids in oncology: An overview. J. Transl. Int. Med. 2014, 2, 78–80. [Google Scholar] [CrossRef]
- Wooldridge, J.E.; Anderson, C.M.; Perry, M.C. Corticosteroids in advanced cancer. Oncology 2001, 15, 225–234. [Google Scholar] [PubMed]
- Lamar, Z.S. The Role of Glucocorticoids in the Treatment of Non-Hodgkin Lymphoma. Ann. Hematol. Oncol. 2016, 3, 1103–1107. [Google Scholar]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol. 2017, 316, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Binnemars-Postma, K.; Storm, G.; Prakash, J. Nanomedicine Strategies to target tumor-associated macrophages. Int. J. Mol. Sci. 2017, 18, 979. [Google Scholar] [CrossRef] [PubMed]
- Banciu, M.; Schiffelers, R.M.; Fens, M.H.; Metselaar, J.M.; Storm, G. Anti-angiogenic effects of liposomal prednisolone phosphate on B16 melanoma in mice. J. Control. Release 2006, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.F.; Jacobs, J.W.; Welsing, P.M.; Verstappen, S.M.; Tekstra, J.; Ton, E.; Geurts, M.A.; van der Werf, J.H.; van Albada-Kuipers, G.A.; Jahangier-de Veen, Z.N.; et al. Low-dose prednisone inclusion in a methotrexate-based, tight control strategy for early rheumatoid arthritis: A randomized trial. Ann. Intern. Med. 2012, 156, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Saag, K.G.; Bridges, S.L., Jr.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016, 68, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Moallem, E.; Koren, E.; Ulmansky, R.; Pizov, G.; Barlev, M.; Barenholz, Y.; Naparstek, Y. A liposomal steroid nano-drug for treating systemic lupus erythematosus. Lupus 2016, 25, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics. 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- La-Beck, N.M.; Gabizon, A.A. Nanoparticle interactions with the immune system: Clinical implications for liposome-based cancer chemotherapy. Front. Immunol. 2017, 8, 416. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Peters, F.M.; Warzecha, K.T.; Bienert, M.; van Bloois, L.; Trautwein, C.; Lammers, T.; Tacke, F. Liposomal encapsulation of dexamethasone modulates cytotoxicity, inflammatory cytokine response, and migratory properties of primary human macrophages. Nanomedicine 2014, 10, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Van der Valk, F.M.; van Wijk, D.F.; Lobatto, M.E.; Verberne, H.J.; Storm, G.; Willems, M.C.; Legemate, D.A.; Nederveen, A.J.; Calcagno, C.; Mani, V.; et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomedicine 2015, 11, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Barrera, P.; Mulder, S.; Smetsers, A.I.; Storm, G.; Beijnen, J.H.; Metselaar, J.M.; van Riel, P.L. Long-circulating liposomal prednisolone versus pulse intramuscular methylprednisolone in patients with active rheumatoid arthritis. Arthritis Rheum. 2008, 58, 3976–3977. [Google Scholar]
- Strehl, C.; van der Goes, M.C.; Bijlsma, J.W.; Jacobs, J.W.; Buttgereit, F. Glucocorticoid-targeted therapies for the treatment of rheumatoid arthritis. Expert Opin. Investig. Drugs 2017, 26, 187–195. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Carrier System | Structural Details | Concerns and Limitations | Medical Applications | References |
|---|---|---|---|---|
| Liposomes and PEGylated liposomes |
|
|
| [34,35,36,37,38,39,40,41,42,43]; [44,45,46,47,48,49,50,51,52,53,54]; [55]; [56,57,58,59,60,61]; [62]; [63,64]; [65] |
| Polymeric micelles |
|
|
| [42]; [50,66] |
| Polymer-drug conjugates |
|
|
| [50,51,67,68,69]; [70]; [71] |
| Inorganic drug delivery systems |
|
|
| [72]; [73]; [74] |
| Inorganic–organic hybrid NP |
|
|
| [75]; Unpublished data; Unpublished data |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lühder, F.; Reichardt, H.M. Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids. Int. J. Mol. Sci. 2017, 18, 1836. https://doi.org/10.3390/ijms18091836
Lühder F, Reichardt HM. Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids. International Journal of Molecular Sciences. 2017; 18(9):1836. https://doi.org/10.3390/ijms18091836
Chicago/Turabian StyleLühder, Fred, and Holger M. Reichardt. 2017. "Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids" International Journal of Molecular Sciences 18, no. 9: 1836. https://doi.org/10.3390/ijms18091836
APA StyleLühder, F., & Reichardt, H. M. (2017). Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids. International Journal of Molecular Sciences, 18(9), 1836. https://doi.org/10.3390/ijms18091836