Natural Killer Cells: Angels and Devils for Immunotherapy

 ,
, 
Abstract
:1. Natural Killer (NK) Cell Modulation Activity
2. NK Cell Classic Cytotoxicity Mechanisms
3. Immunotherapy Strategies Using NK Cells
4. Chimeric Antigen Receptors (CAR) Modified NK Cells
5. CB Derived NK Cells (CB-NK): A Source of Highly Activated NK Cells Which Initiate a Transmissible Cytotoxicity
6. Tumor Cell Survival Mechanisms after Immunotherapy
7. Inflammatory Response: A Double-Edged Sword in Cancer
8. Concluding Remarks
Conflicts of Interest
References
- Morice, W.G. The immunophenotypic attributes of NK cells and NK-cell lineage lymphoproliferative disorders. Am. J. Clin. Pathol. 2007, 127, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Bertaina, A.; Locatelli, F.; Moretta, L. Transplantation and innate immunity: The lesson of natural killer cells. Ital. J. Pediatr. 2009, 35, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Locatelli, F.; Pende, D.; Marcenaro, E.; Mingari, M.C.; Moretta, A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood 2011, 17, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Cooley, S.; Weisdorf, D.J.; Guethlein, L.A.; Klein, J.P.; Wang, T.; Le, C.T.; Marsh, S.G.; Geraghty, D.; Spellman, S.; Haagenson, M.D.; et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 2010, 116, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Feuchtinger, T.; Pfeiffer, M.; Pfaffle, A.; Teltschik, H.M.; Wernet, D.; Schumm, M.; Lotfi, R.; Handgretinger, R.; Lang, P. Cytolytic activity of NK cell clones against acute childhood precursor-B-cell leukaemia is influenced by HLA class I expression on blasts and the differential KIR phenotype of NK clones. Bone Marrow Transplant. 2009, 43, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della Chiesa, M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A.; et al. Markers and function of human NK cells in normal and pathological conditions. Cytom. B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Strauss-Albee, D.M.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Verges, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012, 119, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Verges, S.; Milush, J.M.; Schwartz, B.S.; Pando, M.J.; Jarjoura, J.; York, V.A.; Houchins, J.P.; Miller, S.; Kang, S.M.; Norris, P.J.; et al. Expansion of a unique CD57+ NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 14725–14732. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.; Cooley, S.; Verneris, M.R.; Curtsinger, J.; Luo, X.; Waller, E.K.; Anasetti, C.; Weisdorf, D.; Miller, J.S. Human cytomegalovirus (CMV)-induced memory-like NKG2C+ NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol. 2012, 189, 5082–5088. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Galan, L.; Arenas-Del Angel, M.C.; Zenteno, E.; Chavez, R.; Lascurain, R. Cell death mechanisms induced by cytotoxic lymphocytes. Cell. Mol. Immunol. 2009, 6, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Mace, E.M.; Zhang, J.; Siminovitch, K.A.; Takei, F. Elucidation of the integrin LFA-1-mediated signaling pathway of actin polarization in natural killer cells. Blood 2010, 116, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Susanto, O.; Stewart, S.E.; Voskoboinik, I.; Brasacchio, D.; Hagn, M.; Ellis, S.; Asquith, S.; Sedelies, K.A.; Bird, P.I.; Waterhouse, N.J.; et al. Mouse granzyme A induces a novel death with writhing morphology that is mechanistically distinct from granzyme B-induced apoptosis. Cell Death Differ. 2013, 20, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Joeckel, L.T.; Bird, P.I. Are all granzymes cytotoxic in vivo? Biol. Chem. 2014, 395, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Wang, S.; Zhong, C.; Xue, P.; Fan, Z. Ignition of p53 bomb sensitizes tumor cells to granzyme K-mediated cytolysis. J. Immunol. 2009, 182, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, Q.; Wu, Y.; Wang, L.; Zhou, C.; Ma, W.; Zhang, Y.; Wang, S.; Zhang, S. Granzyme M expressed by tumor cells promotes chemoresistance and EMT in vitro and metastasis in vivo associated with STAT3 activation. Oncotarget 2015, 6, 5818–5831. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, N.J.; Trapani, J.A. H is for helper: Granzyme H helps granzyme B kill adenovirus-infected cells. Trends Immunol. 2007, 28, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.V.; Wilson, C.; Finn, M.W.; Wang, T.; Krensky, A.M.; Clayberger, C. Granulysin delivered by cytotoxic cells damages endoplasmic reticulum and activates caspase-7 in target cells. J. Immunol. 2011, 186, 3497–3504. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G.; et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Cooley, S.; Defor, T.E.; Verneris, M.R.; Zhang, B.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A.; Denman, C.J.; Rondon, G.; Woodworth, G.; Chen, J.; Fisher, T.; Kaur, I.; Fernandez-Vina, M.; Cao, K.; Ciurea, S.; et al. Haploidentical Natural Killer Cells Infused before Allogeneic Stem Cell Transplantation for Myeloid Malignancies: A Phase I Trial. Biol. Blood Marrow Transplant. 2016, 22, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; Parisi, S.; Bontadini, A.; Dan, E.; Motta, M.R.; Rizzi, S.; Trabanelli, S.; Ocadlikova, D.; Lecciso, M.; et al. Larger Size of Donor Alloreactive NK Cell Repertoire Correlates with Better Response to NK Cell Immunotherapy in Elderly Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2016, 22, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Dolstra, H.; Roeven, M.W.H.; Spanholtz, J.; Hangalapura, B.N.; Tordoir, M.; Maas, F.; Leenders, M.; Bohme, F.; Kok, N.; Trilsbeek, C.; et al. Successful Transfer of Umbilical Cord Blood CD34+ Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; le Luduec, J.B.; Forlenza, C.; Jakubowski, A.A.; Perales, M.A.; Young, J.W.; Hsu, K.C. Phase II Study of Haploidentical Natural Killer Cell Infusion for Treatment of Relapsed or Persistent Myeloid Malignancies Following Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Inaba, H.; Kang, G.; Gan, K.; Hartford, C.; Triplett, B.M.; Dallas, M.; Shook, D.; Gruber, T.; Pui, C.H.; et al. Natural killer cell therapy in children with relapsed leukemia. Pediatr. Blood Cancer 2015, 62, 1468–1472. [Google Scholar] [CrossRef] [PubMed]
- Kottaridis, P.D.; North, J.; Tsirogianni, M.; Marden, C.; Samuel, E.R.; Jide-Banwo, S.; Grace, S.; Lowdell, M.W. Two-Stage Priming of Allogeneic Natural Killer Cells for the Treatment of Patients with Acute Myeloid Leukemia: A Phase I Trial. PLoS ONE 2015, 10, e0123416. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; Dupont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br. J. Haematol. 2008, 143, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lim, O.; Kim, T.M.; Ahn, Y.O.; Choi, H.; Chung, H.; Min, B.; Her, J.H.; Cho, S.Y.; Keam, B.; et al. Phase I Study of Random Healthy Donor-Derived Allogeneic Natural Killer Cell Therapy in Patients with Malignant Lymphoma or Advanced Solid Tumors. Cancer Immunol. Res. 2016, 4, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Leivas, A.; Perez-Martinez, A.; Blanchard, M.J.; Martin-Clavero, E.; Fernandez, L.; Lahuerta, J.J.; Martinez-Lopez, J. Novel treatment strategy with autologous activated and expanded natural killer cells plus anti-myeloma drugs for multiple myeloma. Oncoimmunology 2016, 5, e1250051. [Google Scholar] [CrossRef] [PubMed]
- Szmania, S.; Lapteva, N.; Garg, T.; Greenway, A.; Lingo, J.; Nair, B.; Stone, K.; Woods, E.; Khan, J.; Stivers, J.; et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J. Immunother. 2015, 38, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Li, L.; McCarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, A.; Fernandez, L.; Valentin, J.; Martinez-Romera, I.; Corral, M.D.; Ramirez, M.; Abad, L.; Santamaria, S.; Gonzalez-Vicent, M.; Sirvent, S.; et al. A phase I/II trial of interleukin-15–stimulated natural killer cell infusion after haplo-identical stem cell transplantation for pediatric refractory solid tumors. Cytotherapy 2015, 17, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Cryoablation combined with allogenic natural killer cell immunotherapy improves the curative effect in patients with advanced hepatocellular cancer. Oncotarget 2017. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef] [PubMed]
- Barkholt, L.; Alici, E.; Conrad, R.; Sutlu, T.; Gilljam, M.; Stellan, B.; Christensson, B.; Guven, H.; Bjorkstrom, N.K.; Soderdahl, G.; et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: A phase I clinical study. Immunotherapy 2009, 1, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Alici, E.; Sutlu, T.; Bjorkstrand, B.; Gilljam, M.; Stellan, B.; Nahi, H.; Quezada, H.C.; Gahrton, G.; Ljunggren, H.G.; Dilber, M.S. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood 2008, 111, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Sutlu, T.; Stellan, B.; Gilljam, M.; Quezada, H.C.; Nahi, H.; Gahrton, G.; Alici, E. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy 2010, 12, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Granzin, M.; Stojanovic, A.; Miller, M.; Childs, R.; Huppert, V.; Cerwenka, A. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. Oncoimmunology 2016, 5, e1219007. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef] [PubMed]
- Bishara, A.; de Santis, D.; Witt, C.C.; Brautbar, C.; Christiansen, F.T.; Or, R.; Nagler, A.; Slavin, S. The beneficial role of inhibitory KIR genes of HLA class I NK epitopes in haploidentically mismatched stem cell allografts may be masked by residual donor-alloreactive T cells causing GVHD. Tissue Antigens 2004, 63, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.J.; Turner, V.; Handgretinger, R.; Horwitz, E.M.; Benaim, E.; Hale, G.A.; Woodard, P.; Leung, W. T-cell alloreactivity dominates natural killer cell alloreactivity in minimally T-cell-depleted HLA-non-identical paediatric bone marrow transplantation. Br. J. Haematol. 2003, 123, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Martin-Antonio, B.; Yang, H.; Ku, S.; Lee, D.A.; Cooper, L.J.N.; Decker, W.K.; Li, S.; Robinson, S.N.; Sekine, T.; et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS ONE 2013, 8, e76781. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Savoldo, B.; Brenner, M. Fifteen years of gene therapy based on chimeric antigen receptors: “Are we nearly there yet?”. Human Gene Ther. 2009, 20, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent anti-tumor activity. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Fujisaki, H.; Cho, D.; Masselli, M.; Lockey, T.; Eldridge, P.; Leung, W.; Campana, D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy 2012, 14, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Hochberg, J.; Yahr, A.; Ayello, J.; van de Ven, C.; Barth, M.; Czuczman, M.; Cairo, M.S. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol. Res. 2015, 3, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Seidel, D.; Shibina, A.; Siebert, N.; Wels, W.S.; Reynolds, C.P.; Huebener, N.; Lode, H.N. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol. Immunother. 2015, 64, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.; Truscott, L.C.; de Oliveira, S.N. In Vitro Generation of Human NK Cells Expressing Chimeric Antigen Receptor Through Differentiation of Gene-Modified Hematopoietic Stem Cells. Methods Mol. Biol. 2016, 1441, 241–251. [Google Scholar] [PubMed]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Michen, S.; Tietze, S.; Topfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Cantoni, C.; Parolini, S.; Marcenaro, E.; Conte, R.; Moretta, L.; Moretta, A. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur. J. Immunol. 2003, 33, 3439–3447. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56brightCD16− killer Ig-like receptor− NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [PubMed]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef] [PubMed]
- Martin-Antonio, B.; Najjar, A.; Robinson, S.N.; Chew, C.; Li, S.; Yvon, E.; Thomas, M.W.; Mc Niece, I.; Orlowski, R.; Urbano-Ispizua, A.; et al. Transmissible cytotoxicity of Multiple Myeloma cells by NK cells mediated by vesicle trafficking. Cell Death Differ. 2015, 22, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Masilamani, M.; Peruzzi, G.; Borrego, F.; Coligan, J.E. Endocytosis and intracellular trafficking of human natural killer cell receptors. Traffic 2009, 10, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-Garcia, E.; Payer, A.R.; Villa-Alvarez, M.; Fernandez-Guizan, A.; Lopez-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology 2015, 4, e1001224. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Bhattacharya, J. When cells become organelle donors. Physiology 2013, 28, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Khandare, A.; Burianovskyy, L.; Maruyama, S.; Zhang, F.; Nasjletti, A.; Goligorsky, M.S. Tunneling nanotubes mediate rescue of prematurely senescent endothelial cells by endothelial progenitors: Exchange of lysosomal pool. Aging 2011, 3, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Liu, M.; Li, Y.; Nie, Y.; Mi, Q.; Zhao, S. Ovarian tumor-associated microRNA-20a decreases natural killer cell cytotoxicity by downregulating MICA/B expression. Cell. Mol. Immunol. 2014, 11, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kim, J.; Cosman, D.; Choi, I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell. Immunol. 2006, 239, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, M.; Rusakiewicz, S.; Minard-Colin, V.; Delahaye, N.F.; Enot, D.; Vely, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Memmer, S.; Lechner, A.; Huppert, V.; Giannattasio, A.; Becker, T.; Muller-Runte, A.; Lampe, K.; Beutner, D.; Quaas, A.; et al. Natural Killer Group 2D Ligand Depletion Reconstitutes Natural Killer Cell Immunosurveillance of Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2017, 8, 387. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; Dewor, M.; Leng, L.; Bucala, R.; Bernhagen, J. Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74. Cell Signal. 2011, 23, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Krockenberger, M.; Dombrowski, Y.; Weidler, C.; Ossadnik, M.; Honig, A.; Hausler, S.; Voigt, H.; Becker, J.C.; Leng, L.; Steinle, A.; et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J. Immunol. 2008, 180, 7338–7348. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [PubMed]
- Martín-Antonio, B.; Suñe, G.; Najjar, A.; Perez-Amill, L.; Velasco-de Andrés, M.; Lozano, F.; Lozano, E.; Bueno, C.; Estanyol, J.-M.; Muñoz-Pinedo, C.; et al. Natural killer cells transfer antimicrobial and antitumoral Histone H2AZ to kill multiple myeloma cells contributing to transmissible cytotoxicity. Blood 2016, 128, 2115. [Google Scholar]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Das, D.S.; Song, Y.; Richardson, P.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Tallerico, R.; Cristiani, C.M.; Staaf, E.; Garofalo, C.; Sottile, R.; Capone, M.; Pico de Coana, Y.; Madonna, G.; Palella, E.; Wolodarski, M.; et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology 2017, 6, e1261242. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Tramsen, L.; Hanisch, M.; Latge, J.P.; Huenecke, S.; Koehl, U.; Lehrnbecher, T. Human natural killer cells exhibit direct activity against Aspergillus fumigatus hyphae, but not against resting conidia. J. Infect. Dis. 2011, 203, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Sporri, R.; Joller, N.; Albers, U.; Hilbi, H.; Oxenius, A. MyD88-dependent IFN-γ production by NK cells is key for control of Legionella pneumophila infection. J. Immunol. 2006, 176, 6162–6171. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Laorden, M.I.; Stroo, I.; Terpstra, S.; Florquin, S.; Medema, J.P.; de Vos, A.F.; van der Poll, T. Expression and Function of Granzymes A and B in Escherichia coli Peritonitis and Sepsis. Mediat. Inflamm. 2017, 2017, 4137563. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Laorden, M.I.; Stroo, I.; Blok, D.C.; Florquin, S.; Medema, J.P.; de Vos, A.F.; van der Poll, T. Granzymes A and B Regulate the Local Inflammatory Response during Klebsiella pneumoniae Pneumonia. J. Innate Immun. 2016, 8, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Baschuk, N.; Wang, N.; Watt, S.V.; Halse, H.; House, C.; Bird, P.I.; Strugnell, R.; Trapani, J.A.; Smyth, M.J.; Andrews, D.M. NK cell intrinsic regulation of MIP-1α by granzyme M. Cell Death Dis. 2014, 5, e1115. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.M.; Lin, L.C.; Wang, C.F.; Lee, Y.J.; Chen, Y.T.; Liao, Y.D. Antimicrobial Properties of an Immunomodulator—15 kDa Human Granulysin. PLoS ONE 2016, 11, e0156321. [Google Scholar] [CrossRef] [PubMed]
- Stenger, S.; Hanson, D.A.; Teitelbaum, R.; Dewan, P.; Niazi, K.R.; Froelich, C.J.; Ganz, T.; Thoma-Uszynski, S.; Melian, A.; Bogdan, C.; et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 1998, 282, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Wu, T.S.; Hsu, Y.J.; Chang, C.J.; Lin, C.S.; Chia, J.H.; Wu, T.L.; Huang, T.T.; Martel, J.; Ojcius, D.M.; et al. NK cells kill mycobacteria directly by releasing perforin and granulysin. J. Leukoc. Biol. 2014, 96, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Callan, M.F. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheumatol. 2002, 46, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.A.; Andrews, D.M.; Chow, M.; Watt, S.V.; House, C.; Akira, S.; Bird, P.I.; Trapani, J.A.; Smyth, M.J. A role for granzyme M in TLR4-driven inflammation and endotoxicosis. J. Immunol. 2010, 185, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Merkulova, Y.; Raithatha, S.; Parkinson, L.G.; Shen, Y.; Cooper, D.; Granville, D.J. Extracellular granzyme K mediates endothelial activation through the cleavage of protease-activated receptor-1. FEBS J. 2016, 283, 1734–1747. [Google Scholar] [CrossRef] [PubMed]
- Wensink, A.C.; Kemp, V.; Fermie, J.; Garcia Laorden, M.I.; van der Poll, T.; Hack, C.E.; Bovenschen, N. Granzyme K synergistically potentiates LPS-induced cytokine responses in human monocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 5974–5979. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chai, N.R.; Maric, D.; Bielekova, B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J. Immunol. 2011, 187, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Santiago, L.; Menaa, C.; Arias, M.; Martin, P.; Jaime-Sanchez, P.; Metkar, S.; Comas, L.; Erill, N.; Gonzalez-Rumayor, V.; Esser, E.; et al. Granzyme A Contributes to Inflammatory Arthritis in Mice Through Stimulation of Osteoclastogenesis. Arthritis Rheumatol. 2017, 69, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.; Chen, S.; Li, Q.; Lyu, S.C.; Clayberger, C.; Krensky, A.M. Granulysin, a cytolytic molecule, is also a chemoattractant and proinflammatory activator. J. Immunol. 2005, 174, 5243–5248. [Google Scholar] [CrossRef] [PubMed]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, B.C.; Legembre, P.; Pietras, E.; Bubici, C.; Franzoso, G.; Peter, M.E. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004, 23, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Qadir, A.S.; Ceppi, P.; Brockway, S.; Law, C.; Mu, L.; Khodarev, N.N.; Kim, J.; Zhao, J.C.; Putzbach, W.; Murmann, A.E.; et al. CD95/Fas Increases Stemness in Cancer Cells by Inducing a STAT1-Dependent Type I Interferon Response. Cell Rep. 2017, 18, 2373–2386. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell. 2017, 65, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Del Fabbro, E.; Skoro, N.; Brian, C. The prevalence of cachexia among patients with solid and hematologic malignancies at a National Cancer Institute (NCI)-designated cancer center in the 12 months prior to death. J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]



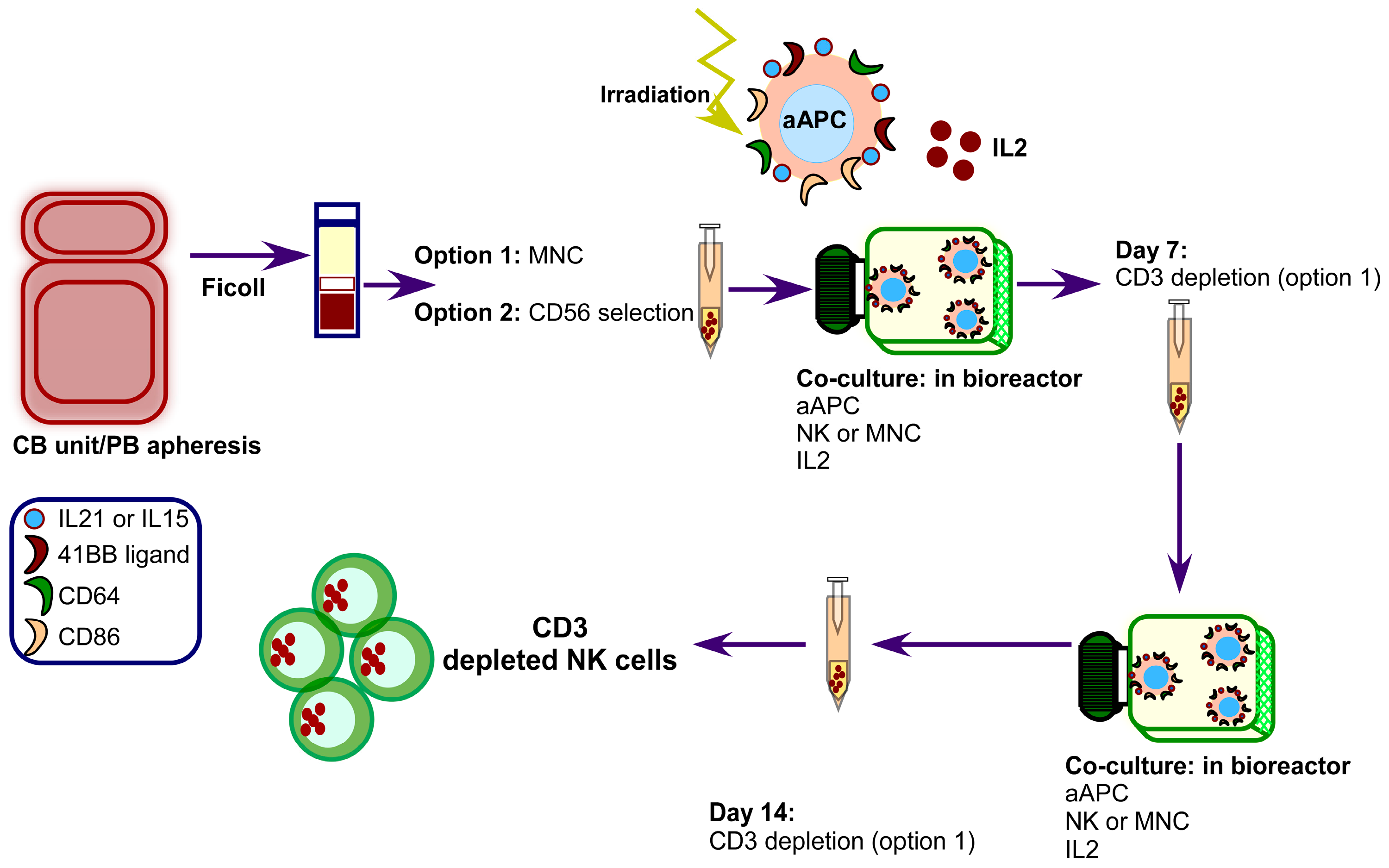{kind=link}
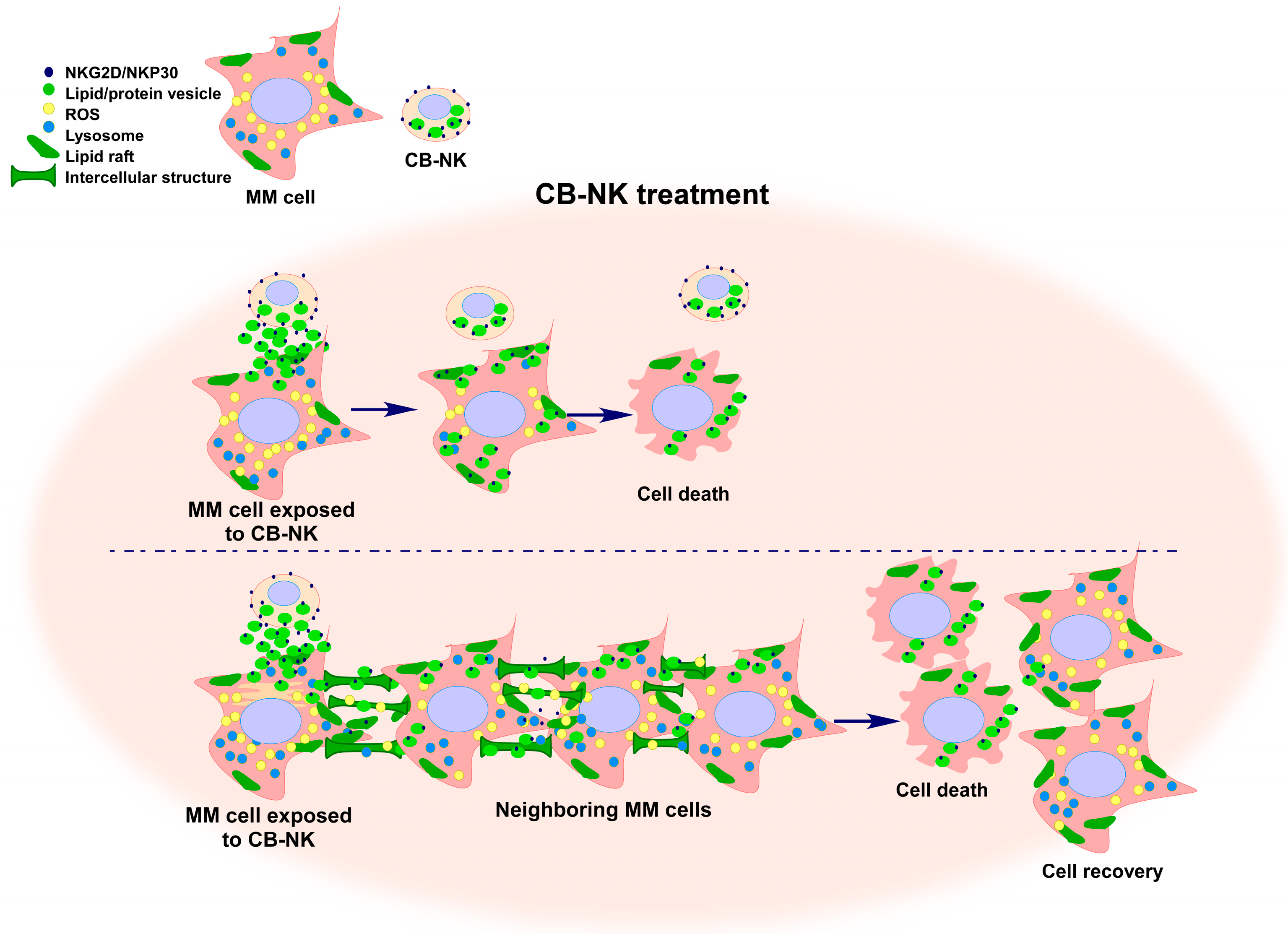{kind=link}
| n Patients Disease | NK Source | Treatment before NK Infusion | NK Activation | Detection of NK in PB | Median Number of Infused NK (×106/Kg) | Graft vs. Host Disease | Outcome. Clinical Trial Number (Reference) |
|---|---|---|---|---|---|---|---|
| 10. AML in CR (pediatric) | Haplo | Flu/Cy | IL2 post-infusion | Yes | 29 | No | All in remission at 964 days [24] |
| 13. AML: 38.4% in AD, 15.3% MR, 46% CR | Haplo | Flu/Cy | IL2 post-infusion | Yes | 2.74 | No | AD: 20% achieved transient CR MR: 100% achieved CR CR: 50% DFS after 34, 32, 18 months. NCT00799799 [25] |
| 16. AML in CR | Haplo | Flu/Cy | IL2 post-infusion | Yes | From 1.29 to 5.53 | No | At 22.5 months: 56% DFS, 44% relapse. Higher NK cell number associated to higher DFS. NCT00799799 [28] |
| 10. AML in CR | Allo NK derived from CD34+ HSPC from CB | Flu/Cy | IL15 and IL2 | Yes (in 21%) | From 3 to 30 | No | 20% became MRD negative for 6 months [29] |
| 57. Refractory AML (15 received IL2DT) | Haplo | Flu/Cy | IL2 post-infusion | Yes: in 10% of patients, and in 27% of patients receiving IL2DT) | 26 | No | CR: 53% (IL2DT) vs. 21% (no IL2DT) DFS: 33% (IL2DT) vs. 5% (no IL2DT) NCT00274846, NCT01106950 [26] |
| 21. AML, MDS, CML | Haplo | Flu, Bu | IL2 pre and post-infusion | NA | From 0.22 to 8.32 | No associated to NK | Survival associated with CD56+ cells delivered; 24% durable CR (no association to KIR-HLA mismatch). NCT00402558. NCT01390402 [27] |
| Refractory 6: AML, 2: MDS | Haplo | Flu/Cy | IL2 post-infusion | No | 10.6 | No | 16% CR; 83% Disease progression. NCT00871689 [30] |
| 29. Pediatric refractory AML Cohort 1: no prior allo-SCT (14) Cohort 2: relapsed after allo-SCT (15) | Haplo | Clo/Eto/Cy | IL2 post-infusion | Yes | From 3.5 to 103 | No | Cohort 1: 71% response; 86% underwent allo-SCT; 36% DFS at 6 years. Cohort 2: 66.6% response and underwent allo-SCT; 27% DFS at 6 years. NCT00697671 and NCT00187096 [31] |
| 7. High risk AML | Haplo | Flu/TBI | Tumor-primed NK cells with tumor lysate | Yes | 3 doses: 1, 5, 10 | No | At 6 months: 42.8% in CR remained in remission, 14% in PR achieved CR, 28% relapse, 14% died. At 1 year: 14% remained in CR. At 2 years: 85.7% died. Median OS: 400 days [32] |
| 10. Relapsed MM | Haplo | Flu/Mel/Dx | IL2 pre and post infusion | Yes (until day 14) | 1.7 | No | 50% CR or near CR, 20% PR, 10% SD and 20% PD [33] |
| 17. Lymphoma (2), advanced solid tumors (15) | Allo | Non immunosuppressive regimen | IL2 (MG4101 method) | Yes | From 1 to 30 (1 and 3 doses) | No | Lymphoma: 50% SD, 50% PD Solid tumors: 47% SD, 53% PD. PFS in SD: 4 months. NCT01212341 [34] |
| 5. Relapsed MM | Auto | Len, Bort | IL2, K562-mb15-41BBL cells | Yes | (7.5)x2 | No | 80% disease stabilization; 40–50% reduction in BM. NCT02481934 [35] |
| 8. Relapsed MM | Auto/Haplo | Bort/Cy/Dx/Flu | K562-mb15-41BBL cells IL2 post-infusion | Yes (in 62%) | 100 | No | 28% partial response [36] |
| 12. Relapsed MM | CB | Len/Mel | K562-mb21-41BBL cells | Yes (in 50%) | 4 doses: 5 , 10, 50 and 100 | No | 83% VGPR, 66% NCR; 33% relapse (at 21 months); 16% dead (at 21 months) [37] |
| 6. Pediatric refractory solid tumors | Haplo | Flu/Bu/Thio/Mp | IL15 | Yes | From 3 to 27 | No | 66% clinical response: 16% VGPR, 33% PR, 16% SD. At 310 days all patients died. NCT01337544 [38] |
| 14. Ovarian 6. Breast | Haplo | Flu/Cy/TBI (in 7 pt) | IL2 pre and post-infusion | In 1 patient (no detection associated to T-reg presence) | 21.6 | No | Toxicity associated to TLS. NCT01105650 [39] |
| 61.Hepatocellular carcinomoa Cryosurgery (26) Cryosurgery+NK (35) | Allo | Cryosurgery | K562-based system | NA | NA | No | Increased PFS: 9.1 vs. 7.6 months Increased Response rate: 60% vs. 46.1% Increased disease control rate: 85.7% vs. 69.2% [40] |
| 7. Metastatic melanoma 1. Renal cell carcinoma | Auto | Flu/Cy | IL2 | Yes | 4.7 | No | 0% response. NCT00328861 [41] |
| 5. CRC (1), .HC (1), RCC (2), CLL (1) | Allo | Ta/Mp (in 2 patients) | IL2 pre and post | Yes | From 1 to 50 | No | 20% PR [42] |
| NCT Number Institution | Type of NK/CAR-Co-Stimulatory Domains | Disease | Treatment/Doses |
|---|---|---|---|
| NCT02892695 PersonGen BioTherapeutics | NK-92 Anti-CD19-CD28, 4-1BB | Relapsed/refractory ALL, CLL, FL, BCL, DLBCL | NK before SCT |
| NCT02944162 PersonGen BioTherapeutics | NK-92 Anti-CD33-CD28, CD137 | Relapsed/refractory CD33+ AML | NK on Days 0, 3 and 5 |
| NCT02742727 PersonGen BioTherapeutics | NK92 Anti-CD7- CD28, 4-1BB | Relapsed/refractory CD7 positive leukemias and lymphomas | NK |
| NCT03056339 M.D.Anderson Cancer Center | CB-NK expanded with K562-mb21 Anti-CD19, 4-1BB, CD28, iCasp9, IL15 | B-cell malignancies: ALL, CLL, NHL | Day-5 to -3: Flu, Cy, Mesna Day 0: NK AP1903 in case of CRS or GVHD |
| NCT02839954 PersonGen BioTherapeutics | NA | Relapsed/refractory Muc1 positive solid tumors | NA |
| NCT01974479 National University Health system, Singapore | Haploidentical NK expanded with K562-mb15-41BBL Anti-CD19, 4-1BB | Refractory ALL | NK |
| NCT00995137 St. Jude Children‘s Research Hospital | Haploidentical NK expanded with K562-mb15-41BBL Anti-CD19, 4-1BB | Refractory ALL | NK |
| Tumor Cell Survival Mechanism | Effect (Reference) |
|---|---|
| Down-regulation of MICA/B and ULBP1/3 (NKG2D ligands) | NK cell inhibition [76,77] |
| Increased levels of soluble ULBP (NKG2D ligand) | NKG2D down-regulation, NK cell inhibition [78] |
| Increased levels of soluble B7-H6 (NKP30 ligand) | NKP30 down-regulation, NK cell inhibition [79] |
| Increased levels of soluble MICA/B (NKG2D ligand) | NK cell inhibition [80] |
| Release of pro-inflammatory molecules (MIF) | NKG2D down-regulation [81,82] |
| Transfer of NKG2D and NKP30 to tumor cells | NKG2D and NKP30 down-regulation in NK cells [69] |
| Up-regulation of inhibitory HLA-G | CAR-NK unresponsive to tumor cells [87] |
| PDL1 over-expression | PD-1 interaction with subsequent NK cell inhibition [90] |
| B7-H3 over-expression | NK cell inhibition [91] |
| TIM-3 over-expression | NK cell inhibition [93] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Antonio, B.; Suñe, G.; Perez-Amill, L.; Castella, M.; Urbano-Ispizua, A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int. J. Mol. Sci. 2017, 18, 1868. https://doi.org/10.3390/ijms18091868
Martín-Antonio B, Suñe G, Perez-Amill L, Castella M, Urbano-Ispizua A. Natural Killer Cells: Angels and Devils for Immunotherapy. International Journal of Molecular Sciences. 2017; 18(9):1868. https://doi.org/10.3390/ijms18091868
Chicago/Turabian StyleMartín-Antonio, Beatriz, Guillermo Suñe, Lorena Perez-Amill, Maria Castella, and Alvaro Urbano-Ispizua. 2017. "Natural Killer Cells: Angels and Devils for Immunotherapy" International Journal of Molecular Sciences 18, no. 9: 1868. https://doi.org/10.3390/ijms18091868
APA StyleMartín-Antonio, B., Suñe, G., Perez-Amill, L., Castella, M., & Urbano-Ispizua, A. (2017). Natural Killer Cells: Angels and Devils for Immunotherapy. International Journal of Molecular Sciences, 18(9), 1868. https://doi.org/10.3390/ijms18091868