The Complex Interplay between DNA Injury and Repair in Enzymatically Induced Mutagenesis and DNA Damage in B Lymphocytes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
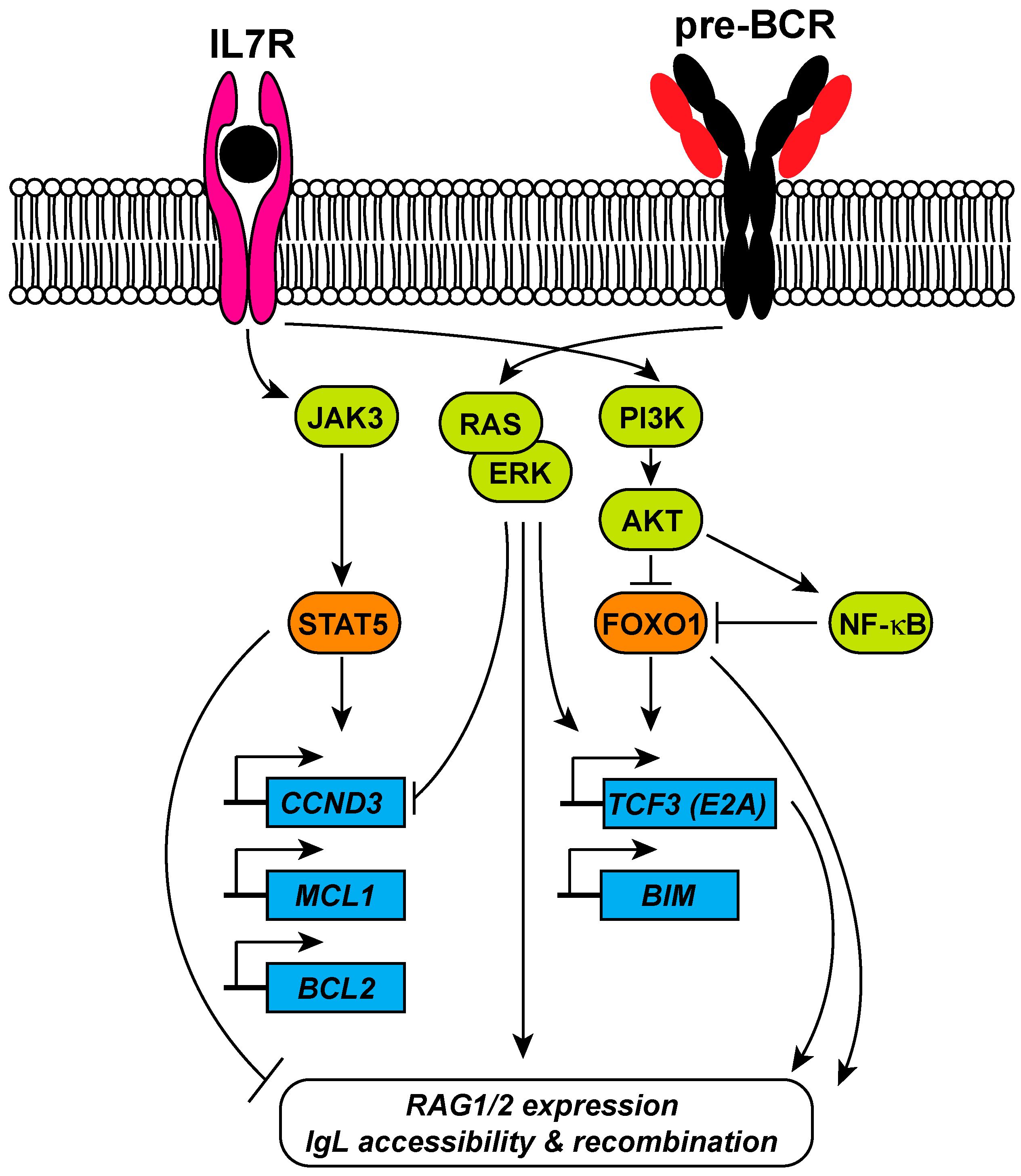{kind=link}
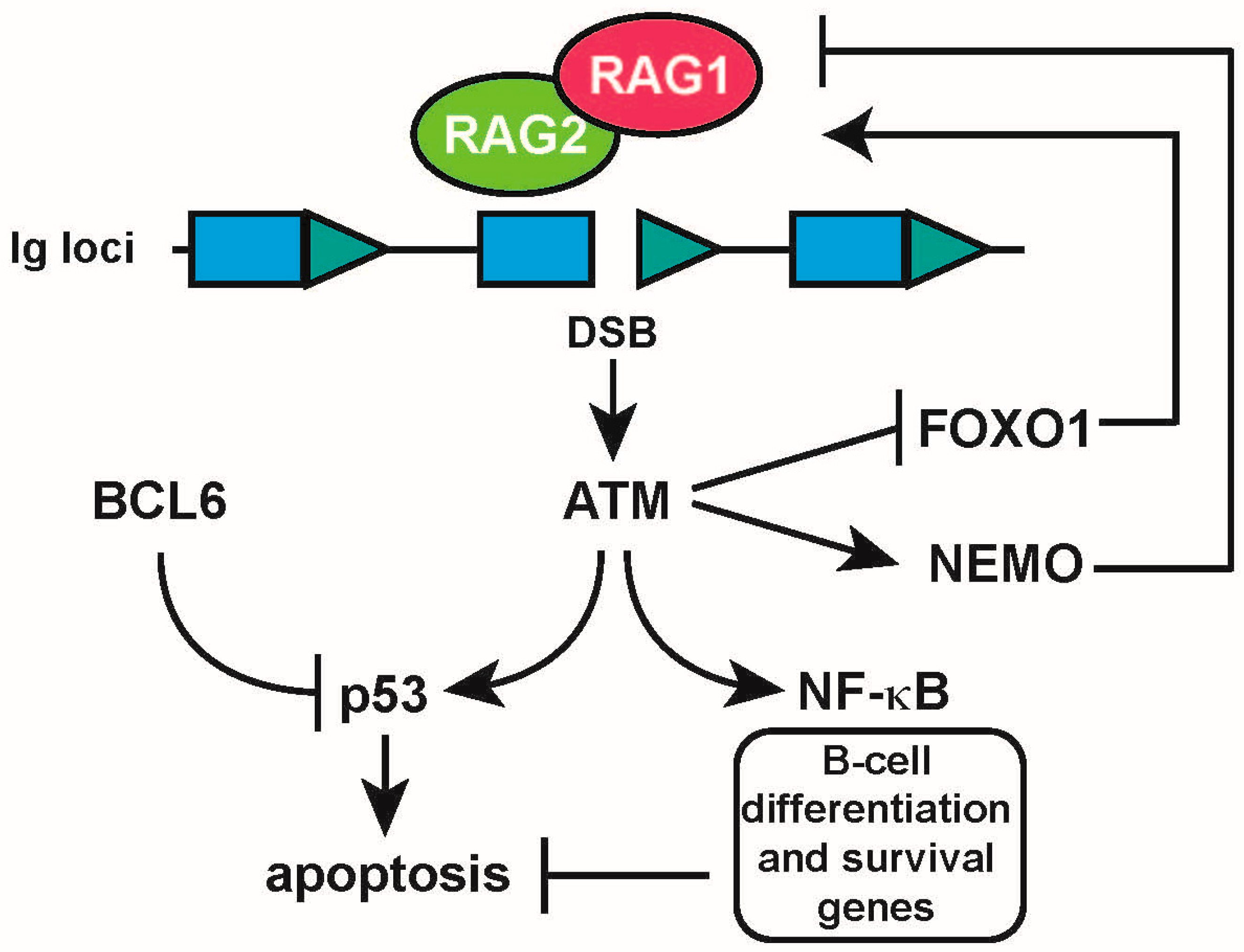{kind=link}
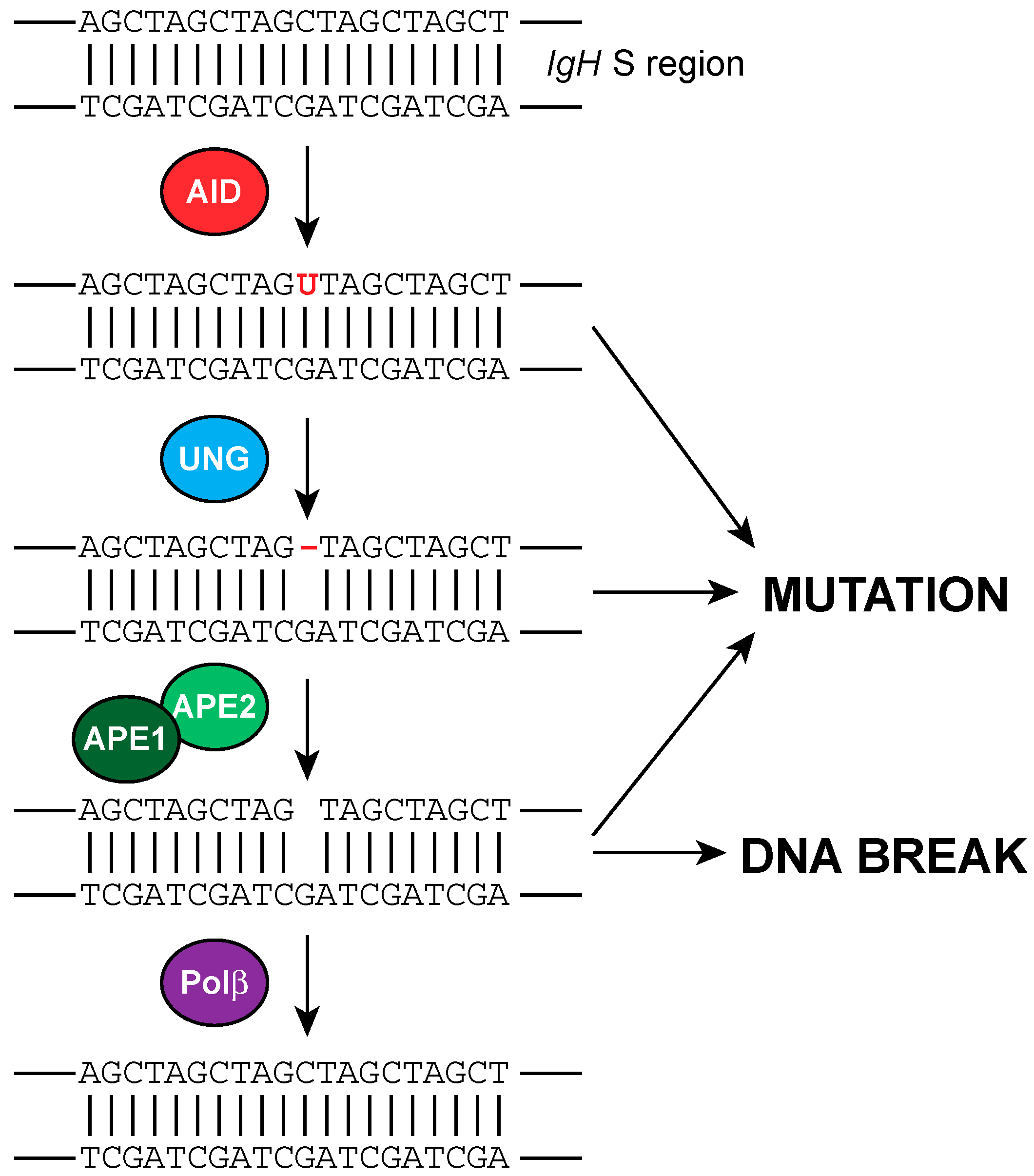{kind=link}
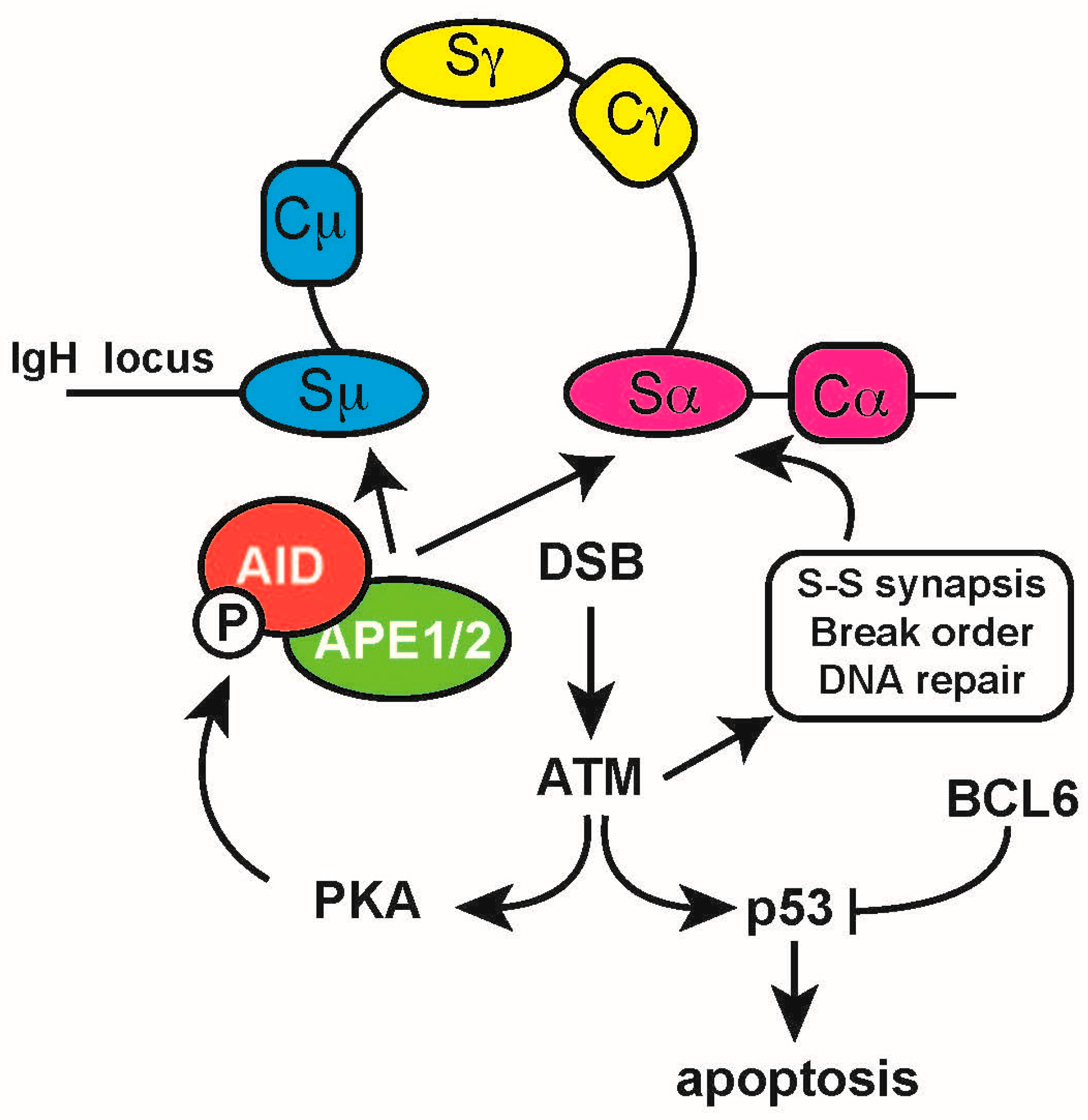{kind=link}
{kind=link}
{kind=link}
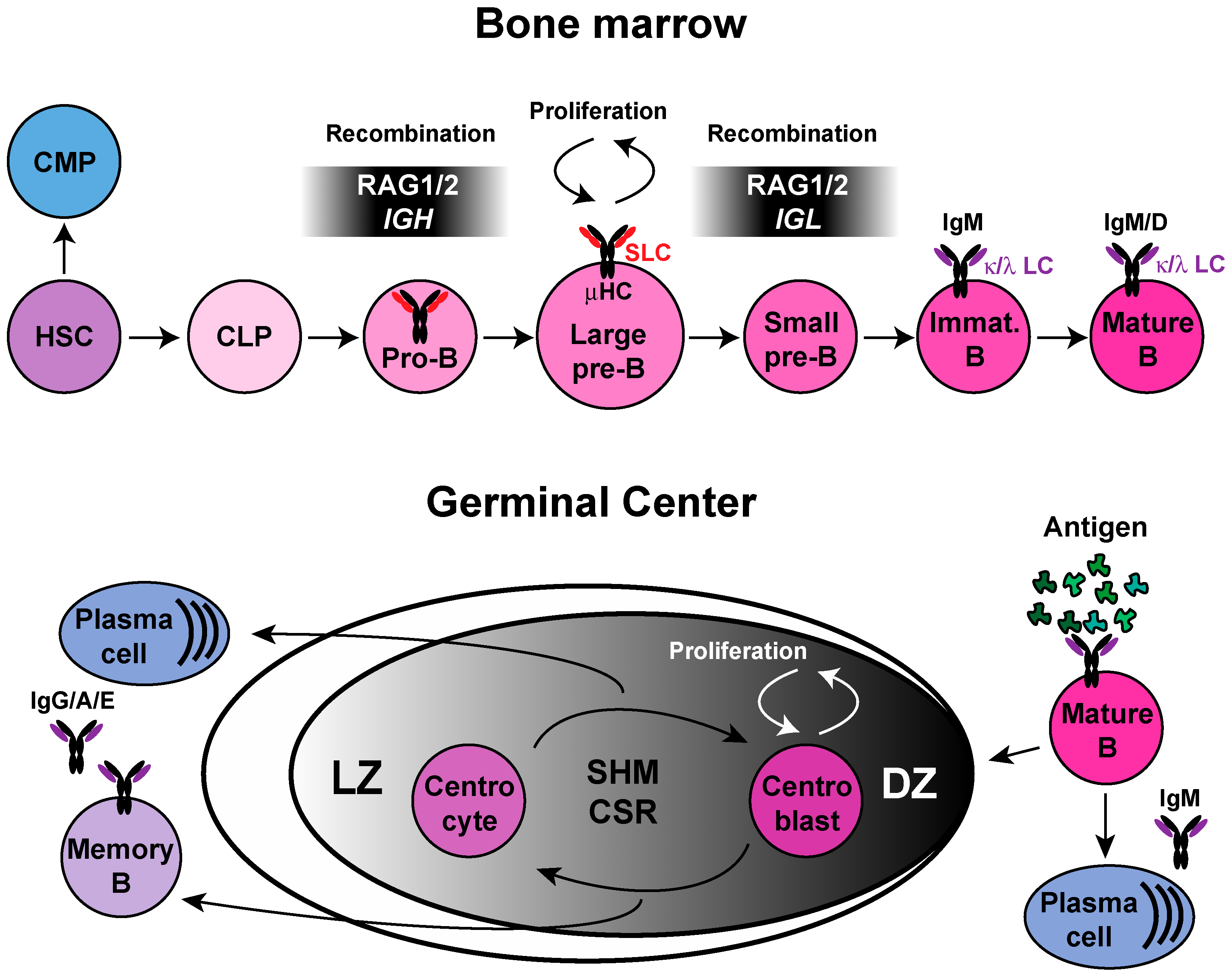
1. B-Cell Development
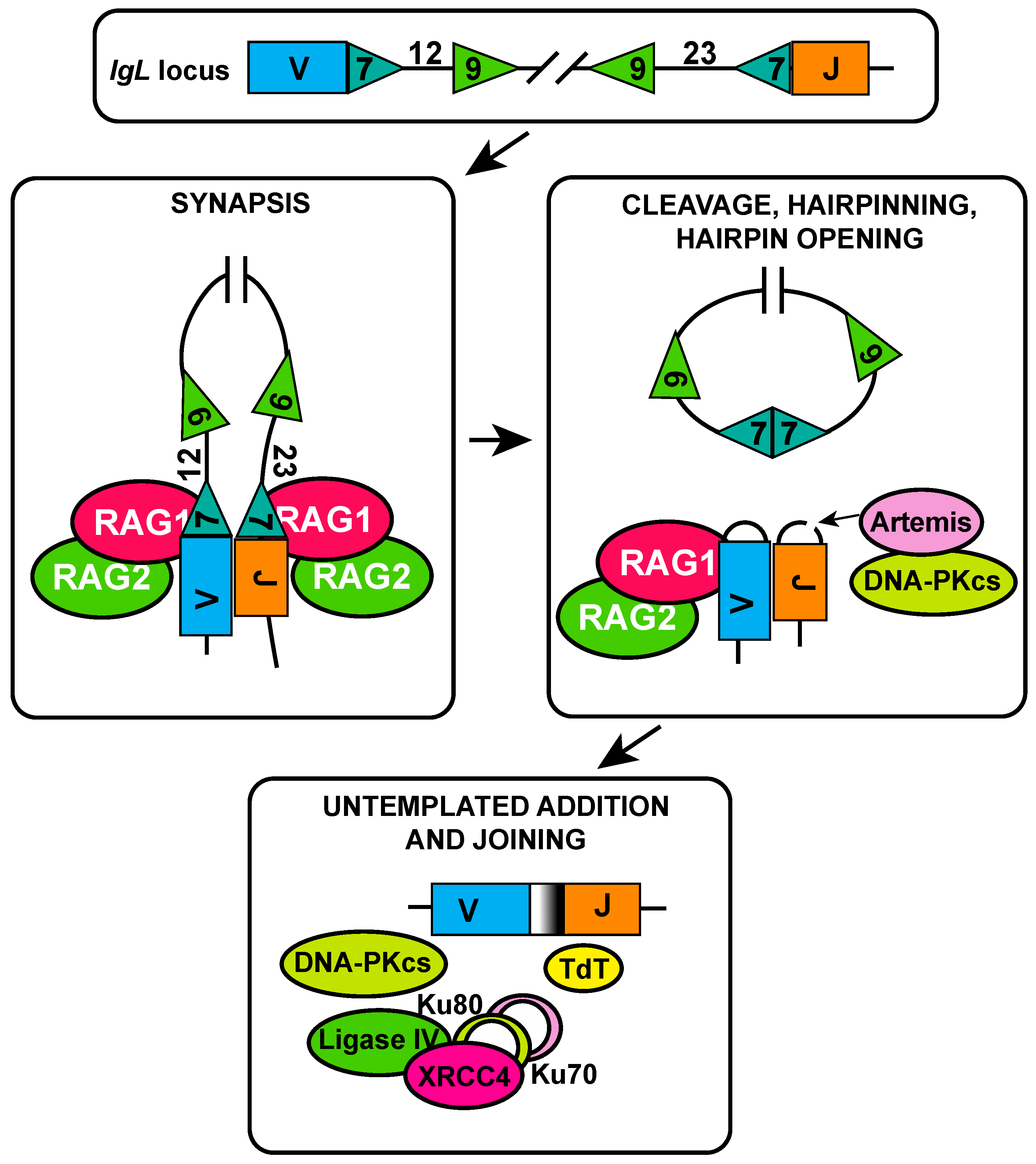
2. RAG and V(D)J Recombination
3. Regulation of RAG1 and RAG2 Expression and Activity
4. Signaling Pathways Involved in RAG1 and RAG2 Expression
5. The DNA Damage Response Regulates RAG Activity
6. Malignancies Associated with Dysregulated RAG Activity
7. The Mechanism of Class Switch Recombination and Involvement of Base Excision Repair (BER)
8. Role of Mismatch Repair Factors in Conversion of SSBs to DSBs
9. DNA Damage Response Regulates CSR and AID
10. CSR and Chromosomal Translocations
11. Antibody Diversification by SHM
12. Involvement of BER and MMR in SHM
13. Aberrant SHM Activity and its Contribution to Genomic Instability
14. DDR Regulation in the Germinal Center
15. AID-Induced Localized Hypermutations and AID Activity in Other Cell Types
16. Concluding Remarks and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hardy, R.R.; Hayakawa, K. B cell development pathways. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T. Microenvironmental niches in the bone marrow required for B-cell development. Nat. Rev. Immunol. 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Kee, B.L. The transcriptional regulation of B cell lineage commitment. Immunity 2007, 26, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Roessler, S.; Grosschedl, R. Role of transcription factors in commitment and differentiation of early B lymphoid cells. Semin. Immunol. 2006, 18, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Dorshkind, K. Understanding how pre-B cells come of age. Nat. Immunol. 2000, 1, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, R. The Development of B Cells in the Bone Marrow Is Controlled by the Balance between Cell-Autonomous Mechanisms and Signals from the Microenvironment. J. Exp. Med. 2000, 191, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Grundy, G.J.; Ramon-Maiques, S.; Dimitriadis, E.K.; Kotova, S.; Biertumpfel, C.; Heymann, J.B.; Steven, A.C.; Gellert, M.; Yang, W. Initial stages of V(D)J recombination: The organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol. Cell 2009, 35, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Oettinger, M.A.; Schatz, D.G.; Gorka, C.; Baltimore, D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 1990, 248, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.G.; Oettinger, M.A.; Baltimore, D. The V(D)J recombination activating gene, RAG-1. Cell 1989, 59, 1035–1048. [Google Scholar] [CrossRef]
- Tiegs, S.L.; Russell, D.M.; Nemazee, D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993, 177, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Vettermann, C.; Schlissel, M.S. Allelic exclusion of immunoglobulin genes: Models and mechanisms. Immunol. Rev. 2010, 237, 22–42. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, K.; Forster, I.; Cumano, A. Evolutionary and somatic selection of the antibody repertoire in the mouse. Science 1987, 238, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Oropallo, M.A.; Cerutti, A. Germinal center reaction: Antigen affinity and presentation explain it all. Trends Immunol. 2014, 35, 287–289. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, D.C.; Ramsden, D.A.; Gellert, M. The RAG1 and RAG2 Proteins Establish the 12/23 Rule in V(D)J Recombination. Cell 1996, 85, 107–113. [Google Scholar] [CrossRef]
- Eastman, Q.M.; Leu, T.M.; Schatz, D.G. Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature 1996, 380, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Rooney, S.; Sekiguchi, J.; Zhu, C.; Cheng, H.L.; Manis, J.; Whitlow, S.; DeVido, J.; Foy, D.; Chaudhuri, J.; Lombard, D.; Alt, F.W. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol. Cell 2002, 10, 1379–1390. [Google Scholar] [CrossRef]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar] [PubMed]
- Roth, D.B. V(D)J Recombination: Mechanism, Errors, and Fidelity. Microbiol. Spectr. 2014. [CrossRef] [PubMed]
- Fugmann, S.D.; Lee, A.I.; Shockett, P.E.; Villey, I.J.; Schatz, D.G. The RAG proteins and V(D)J recombination: Complexes, ends, and transposition. Annu. Rev. Immunol. 2000, 18, 495–527. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 2004, 3, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Lescale, C.; Lenden Hasse, H.; Blackford, A.N.; Balmus, G.; Bianchi, J.J.; Yu, W.; Bacoccina, L.; Jarade, A.; Clouin, C.; Sivapalan, R.; et al. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep. 2016, 16, 2967–2979. [Google Scholar] [CrossRef] [PubMed]
- Savarese, F.; Grosschedl, R. FOXtrot and RAGtime in B cells. Nat. Immunol. 2006, 7, 793–794. [Google Scholar] [CrossRef] [PubMed]
- Lauring, J.; Schlissel, M.S. Distinct Factors Regulate the Murine RAG-2 Promoter in B- and T-Cell Lines. Mol. Cell. Biol. 1999, 19, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Misulovin, Z.; Suh, H.; Hardy, R.R.; Jankovic, M.; Yannoutsos, N.; Nussenzweig, M.C. Coordinate regulation of RAG1 and RAG2 by cell type-specific DNA elements 5′ of RAG2. Science 1999, 285, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Kishi, H.; Jin, Z.X.; Wei, X.C.; Nagata, T.; Matsuda, T.; Saito, S.; Muraguchi, A. Cooperative binding of c-Myb and Pax-5 activates the RAG-2 promoter in immature B cells. Blood 2002, 99, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.X.; Kishi, H.; Wei, X.C.; Matsuda, T.; Saito, S.; Muraguchi, A. Lymphoid enhancer-binding factor-1 binds and activates the recombination-activating gene-2 promoter together with c-Myb and Pax-5 in immature B cells. J. Immunol. 2002, 169, 3783–3792. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.Y.; Lauring, J.; Liang, H.E.; Greenbaum, S.; Cado, D.; Zhuang, Y.; Schlissel, M.S. A conserved transcriptional enhancer regulates RAG gene expression in developing B cells. Immunity 2003, 19, 105–117. [Google Scholar] [CrossRef]
- Kuo, T.C.; Schlissel, M.S. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr. Opin. Immunol. 2009, 21, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Desiderio, S. Cyclin A/CDK2 regulates V(D)J recombination by coordinating RAG-2 accumulation and DNA repair. Immunity 1999, 11, 771–781. [Google Scholar] [CrossRef]
- Jiang, H.; Chang, F.C.; Ross, A.E.; Lee, J.; Nakayama, K.; Nakayama, K.; Desiderio, S. Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Mol. Cell 2005, 18, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Cobb, R.M.; Oestreich, K.J.; Osipovich, O.A.; Oltz, E.M. Accessibility control of V(D)J recombination. Adv. Immunol. 2006, 91, 45–109. [Google Scholar] [PubMed]
- Stanhope-Baker, P.; Hudson, K.M.; Shaffer, A.L.; Constantinescu, A.; Schlissel, M.S. Cell type-specific chromatin structure determines the targeting of V(D)J recombinase activity in vitro. Cell 1996, 85, 887–897. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Desiderio, S. Temporal and spatial regulation of V(D)J recombination: Interactions of extrinsic factors with the RAG complex. Adv. Exp. Med. Biol. 2009, 650, 157–165. [Google Scholar] [PubMed]
- Ramón-Maiques, S.; Kuo, A.J.; Carney, D.; Matthews, A.G.W.; Oettinger, M.A.; Gozani, O.; Yang, W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc. Natl. Acad. Sci. USA 2007, 104, 18993–18998. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Subrahmanyam, R.; Chakraborty, T.; Sen, R.; Desiderio, S. A Plant Homeodomain in Rag-2 that Binds Hypermethylated Lysine 4 of Histone H3 Is Necessary for Efficient Antigen-Receptor-Gene Rearrangement. Immunity 2007, 27, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reynolds, T.L.; Shan, X.; Desiderio, S. Coupling of V(D)J recombination to the cell cycle suppresses genomic instability and lymphoid tumorigenesis. Immunity 2011, 34, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R.; Mandal, M.; Ochiai, K.; Singh, H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 2014, 14, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Grawunder, U.; Leu, T.M.; Schatz, D.G.; Werner, A.; Rolink, A.G.; Melchers, F.; Winkler, T.H. Down-regulation of RAG1 and RAG2 gene expression in preB cells after functional immunoglobulin heavy chain rearrangement. Immunity 1995, 3, 601–608. [Google Scholar] [CrossRef]
- Malin, S.; McManus, S.; Cobaleda, C.; Novatchkova, M.; Delogu, A.; Bouillet, P.; Strasser, A.; Busslinger, M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat. Immunol. 2010, 11, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Herzog, S.; Reth, M.; Jumaa, H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat. Rev. Immunol. 2009, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, K.; Maienschein-Cline, M.; Mandal, M.; Triggs, J.R.; Bertolino, E.; Sciammas, R.; Dinner, A.R.; Clark, M.R.; Singh, H. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat. Immunol. 2012, 13, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Scherer, D.C.; Brockman, J.A.; Bendall, H.H.; Zhang, G.M.; Ballard, D.W.; Oltz, E.M. Corepression of RelA and c-rel inhibits immunoglobulin kappa gene transcription and rearrangement in precursor B lymphocytes. Immunity 1996, 5, 563–574. [Google Scholar] [CrossRef]
- Sasaki, Y.; Derudder, E.; Hobeika, E.; Pelanda, R.; Reth, M.; Rajewsky, K.; Schmidt-Supprian, M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 2006, 24, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Derudder, E.; Cadera, E.J.; Vahl, J.C.; Wang, J.; Fox, C.J.; Zha, S.; van Loo, G.; Pasparakis, M.; Schlissel, M.S.; Schmidt-Supprian, M.; Rajewsky, K. Development of immunoglobulin lambda-chain-positive B cells, but not editing of immunoglobulin kappa-chain, depends on NF-kappaB signals. Nat. Immunol. 2009, 10, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, H.; Baba, Y.; Nagai, Y.; Jimi, E.; Ghosh, S.; Kincade, P.W. NF-kappaB is dispensable for normal lymphocyte development in bone marrow but required for protection of progenitors from TNFalpha. Int. Immunol. 2006, 18, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Cadera, E.J.; Wan, F.; Amin, R.H.; Nolla, H.; Lenardo, M.J.; Schlissel, M.S. NF-kappaB activity marks cells engaged in receptor editing. J. Exp. Med. 2009, 206, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Verkoczy, L.; Aїt-Azzouzene, D.; Skog, P.; Märtensson, A.; Lang, J.; Duong, B.; Nemazee, D. A Role for Nuclear Factor Kappa B/Rel Transcription Factors in the Regulation of the Recombinase Activator Genes. Immunity 2005, 22, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Balkhi, M.Y.; Willette-Brown, J.; Zhu, F.; Chen, Z.; Liu, S.; Guttridge, D.C.; Karin, M.; Hu, Y. IKKalpha-mediated signaling circuitry regulates early B lymphopoiesis during hematopoiesis. Blood 2012, 119, 5467–5477. [Google Scholar] [CrossRef] [PubMed]
- Ochodnicka-Mackovicova, K.; Bahjat, M.; Bloedjes, T.A.; Maas, C.; de Bruin, A.M.; Bende, R.J.; van Noesel, C.J.M.; Guikema, J.E. NF-κB and AKT signaling prevent DNA damage in transformed pre-B cells by suppressing RAG1/2 expression and activity. Blood 2015, 126, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y. CDK4 deficiency promotes genomic instability and enhances. J. Clin. Investig. 2014, 124, 1672–1684. [Google Scholar] [PubMed]
- Li, J.; Joo, S.H.; Tsai, M.D. An NF-kappaB-specific inhibitor, IkappaBalpha, binds to and inhibits cyclin-dependent kinase 4. Biochemistry 2003, 42, 13476–13483. [Google Scholar] [CrossRef] [PubMed]
- Durocher, D.; Jackson, S.P. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr. Opin. Cell. Biol. 2001, 13, 225–231. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Bredemeyer, A.L.; Sharma, G.G.; Huang, C.-Y.; Helmink, B.A.; Walker, L.M.; Khor, K.C.; Nuskey, B.; Sullivan, K.E.; Pandita, T.K.; Bassing, C.H.; et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006, 442, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Callen, E.; Jankovic, M.; Difilippantonio, S.; Daniel, J.A.; Chen, H.T.; Celeste, A.; Pellegrini, M.; McBride, K.; Wangsa, D.; Bredemeyer, A.L.; et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 2007, 130, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.L.; Yin, B.; Ji, Y.; Chaumeil, J.; Marszalek, K.; Tenthorey, J.; Salvagiotto, G.; Steinel, N.; Ramsey, L.B.; Ghysdael, J.; et al. RAG-1 and ATM coordinate monoallelic recombination and nuclear positioning of immunoglobulin loci. Nat. Immunol. 2009, 10, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Steinel, N.C.; Fisher, M.R.; Yang-Iott, K.S.; Bassing, C.H. The Ataxia Telangiectasia Mutated and Cyclin D3 Proteins Cooperate to Help Enforce TCRβ and IgH Allelic Exclusion. J. Immunol. 2014, 193, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Hiom, K.; Gellert, M. A Stable RAG1-RAG2-DNA Complex That Is Active in V(D)J Cleavage. Cell 1997, 88, 65–72. [Google Scholar] [CrossRef]
- Taylor, A.M.; Metcalfe, J.A.; Thick, J.; Mak, Y.F. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996, 87, 423–438. [Google Scholar] [PubMed]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef]
- Liyanage, M.; Weaver, Z.; Barlow, C.; Coleman, A.; Pankratz, D.G.; Anderson, S.; Wynshaw-Boris, A.; Ried, T. Abnormal rearrangement within the alpha/delta T-cell receptor locus in lymphomas from Atm-deficient mice. Blood 2000, 96, 1940–1946. [Google Scholar] [PubMed]
- Xu, Y.; Ashley, T.; Brainerd, E.E.; Bronson, R.T.; Meyn, M.S.; Baltimore, D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996, 10, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.J.; Van Dyke, T. Critical role for Atm in suppressing V(D)J recombination-driven thymic lymphoma. Genes Dev. 1999, 13, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Ochodnicka-Mackovicova, K.; Bahjat, M.; Maas, C.; van der Veen, A.; Bloedjes, T.A.; de Bruin, A.M.; van Andel, H.; Schrader, C.E.; Hendriks, R.W.; Verhoeyen, E.; et al. The DNA Damage Response Regulates RAG1/2 Expression in Pre-B Cells through ATM-FOXO1 Signaling. J. Immunol. 2016, 197, 2918–2929. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.R.; Rivera-Reyes, A.; Bloch, N.B.; Schatz, D.G.; Bassing, C.H. Immature Lymphocytes Inhibit Rag1 and Rag2 Transcription and V(D)J Recombination in Response to DNA Double-Strand Breaks. J. Immunol. 2017, 198, 2943–2956. [Google Scholar] [CrossRef] [PubMed]
- Bredemeyer, A.L.; Helmink, B.A.; Innes, C.L.; Calderon, B.; McGinnis, L.M.; Mahowald, G.K.; Gapud, E.J.; Walker, L.M.; Collins, J.B.; Weaver, B.K.; et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature 2008, 456, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Duy, C.; Yu, J.J.; Nahar, R.; Swaminathan, S.; Kweon, S.M.; Polo, J.M.; Valls, E.; Klemm, L.; Shojaee, S.; Cerchietti, L.; et al. BCL6 is critical for the development of a diverse primary B cell repertoire. J. Exp. Med. 2010, 207, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.V.R.; Perkins, E.J.; Ramsden. Genomic instability due to V(D)J recombination-associated transposition. Genes Dev. 2006, 20, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 2016, 16, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Pepin, F.; Carlton, V.E.H.; Klinger, M.; Logan, A.C.; Miklos, D.B.; Faham, M.; Dahl, G.; Lacayo, N. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood 2012, 120, 4407–4417. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Rosenwald, A.; Staudt, L.M. Lymphoid malignancies: The dark side of B-cell differentiation. Nat. Rev. Immunol. 2002, 2, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Gladdy, R.A.; Taylor, M.D.; Williams, C.J.; Grandal, I.; Karaskova, J.; Squire, J.A.; Rutka, J.T.; Guidos, C.J.; Danska, J.S. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell 2003, 3, 37–50. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; Potter, N.E.; Wedge, D.C.; Tubio, J.; Alexandrov, L.B.; Van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, E.J.; Verhagen, O.J.; van Leeuwen, E.F.; von dem Borne, A.E.; van der Schoot, C.E. Distinct ongoing Ig heavy chain rearrangement processes in childhood B-precursor acute lymphoblastic leukemia. Blood 1993, 82, 581–589. [Google Scholar] [PubMed]
- Stavnezer, J.; Guikema, J.E.; Schrader, C.E. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008, 26, 261–292. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.E.; Linehan, E.K.; Mochegova, S.N.; Woodland, R.T.; Stavnezer, J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J. Exp. Med. 2005, 202, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Casellas, R.; Reina-San-Martin, B.; Chen, H.T.; Difilippantonio, M.J.; Wilson, P.C.; Hanitsch, L.; Celeste, A.; Muramatsu, M.; Pilch, D.R.; et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature 2001, 414, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Schrader, C.E. Ig heavy chain class switch recombination: Mechanism and regulation. J. Immunol. 2014, 193, 5370–5378. [Google Scholar] [CrossRef] [PubMed]
- Rada, C.; Williams, G.T.; Nilsen, H.; Barnes, D.E.; Lindahl, T.; Neuberger, M.S. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002, 12, 1748–1755. [Google Scholar] [CrossRef]
- Imai, K.; Slupphaug, G.; Lee, W.I.; Revy, P.; Nonoyama, S.; Catalan, N.; Yel, L.; Forveille, M.; Kavli, B.; Krokan, H.E.; et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003, 4, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Di Noia, J.; Neuberger, M.S. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 2002, 419, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Begum, N.A.; Stanlie, A.; Doi, T.; Sasaki, Y.; Jin, H.W.; Kim, Y.S.; Nagaoka, H.; Honjo, T. Further evidence for involvement of a noncanonical function of uracil DNA glycosylase in class switch recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 2752–2757. [Google Scholar] [CrossRef] [PubMed]
- Stivers, J.T. Comment on “Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch”. Science 2004. [Google Scholar] [CrossRef] [PubMed]
- Dingler, F.A.; Kemmerich, K.; Neuberger, M.S.; Rada, C. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A:T substitutions during somatic mutation. Eur. J. Immunol. 2014, 44, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Grigera, F.; Wuerffel, R.; Kenter, A.L. MBD4 Facilitates Immunoglobulin Class Switch Recombination. Mol. Cell. Biol. 2017. [CrossRef] [PubMed]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013. [CrossRef] [PubMed]
- Fung, H.; Demple, B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 2005, 17, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Guikema, J.E.J.; Linehan, E.K.; Tsuchimoto, D.; Nakabeppu, Y.; Strauss, P.R.; Stavnezer, J.; Schrader, C.E. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J. Exp. Med. 2007, 204, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Meira, L.B.; Devaraj, S.; Kisby, G.E.; Burns, D.K.; Daniel, R.L.; Hammer, R.E.; Grundy, S.; Jialal, I.; Friedberg, E.C. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer res. 2001, 61, 5552–5557. [Google Scholar] [PubMed]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 protein has a 3′–5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006, 34, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Alili, D.; Saint-Pierre, C.; Gasparutto, D.; Zharkov, D.O.; Ishchenko, A.A.; Tudek, B.; Saparbaev, M.K. Uracil in duplex DNA is a substrate for the nucleotide incision repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3695–E3703. [Google Scholar] [CrossRef] [PubMed]
- Ide, Y.; Tsuchimoto, D.; Tominaga, Y.; Nakashima, M.; Watanabe, T.; Sakumi, K.; Ohno, M.; Nakabeppu, Y. Growth retardation and dyslymphopoiesis accompanied by G2/M arrest in APEX2-null mice. Blood 2004, 104, 4097–4103. [Google Scholar] [CrossRef] [PubMed]
- Hadi, M.Z.; Ginalski, K.; Nguyen, L.H.; Wilson, D.M., 3rd. Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J. Mol. Biol. 2002, 316, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.E.; Guikema, J.E.; Wu, X.; Stavnezer, J. The roles of APE1, APE2, DNA polymerase beta and mismatch repair in creating S region DNA breaks during antibody class switch. Philos. Trans. R. Soc. Biol. Sci. 2009, 364, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Husain, A.; Hu, W.; Honjo, T.; Kobayashi, M. APE1 is dispensable for S-region cleavage but required for its repair in class switch recombination. Proc. Natl. Acad. Sci. USA 2014, 111, 17242–17247. [Google Scholar] [CrossRef] [PubMed]
- Masani, S.; Han, L.; Yu, K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol. Cell Biol. 2013, 33, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Stavnezer, J. DNA polymerase β is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J. Exp. Med. 2007, 204, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.E.; Vardo, J.; Stavnezer, J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J. Exp. Med. 2002, 195, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Scherer, S.J.; Ronai, D.; Iglesias-Ussel, M.D.; Peled, J.U.; Bardwell, P.D.; Zhuang, M.; Lee, K.; Martin, A.; Edelmann, W.; Scharff, M.D. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. J. Exp. Med. 2004, 200, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.E.; Guikema, J.E.; Linehan, E.K.; Selsing, E.; Stavnezer, J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J. Immunol. 2007, 179, 6064–6071. [Google Scholar] [CrossRef] [PubMed]
- Min, I.M.; Schrader, C.E.; Vardo, J.; Luby, T.M.; D’Avirro, N.; Stavnezer, J.; Selsing, E. The Smu tandem repeat region is critical for Ig isotype switching in the absence of Msh2. Immunity 2003, 19, 515–524. [Google Scholar] [CrossRef]
- Eccleston, J.; Yan, C.; Yuan, K.; Alt, F.W.; Selsing, E. Mismatch repair proteins MSH2, MLH1, and EXO1 are important for class-switch recombination events occurring in B cells that lack nonhomologous end joining. J. Immunol. 2011, 186, 2336–2343. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Schrader, C.E. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. 2006, 22, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Guikema, J.E.; Schrader, C.E.; Leus, N.G.; Ucher, A.; Linehan, E.K.; Werling, U.; Edelmann, W.; Stavnezer, J. Reassessment of the role of Mut S homolog 5 in Ig class switch recombination shows lack of involvement in cis- and trans-switching. J. Immunol. 2008, 181, 8450–8459. [Google Scholar] [CrossRef] [PubMed]
- Staszewski, O.; Baker, R.E.; Ucher, A.J.; Martier, R.; Stavnezer, J.; Guikema, J.E. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol. Cell 2011, 41, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, J.M.; McCarty, T.; Petiniot, L.K.; Shen, R.; Barlow, C.; Wynn, T.A.; Morse, H.C., 3rd; Gearhart, P.J.; Wynshaw-Boris, A.; Max, E.E.; et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J. Exp. Med. 2004, 200, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Reina-San-Martin, B.; Chen, H.T.; Nussenzweig, A.; Nussenzweig, M.C. ATM is required for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 2004, 200, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Reina-San-Martin, B.; Olaru, A.; Minn, K.; Tamada, K.; Lau, J.S.; Cascalho, M.; Chen, L.; Nussenzweig, A.; Livak, F.; et al. 53BP1 is required for class switch recombination. J. Cell Biol. 2004, 165, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Manis, J.P.; Morales, J.C.; Xia, Z.; Kutok, J.L.; Alt, F.W.; Carpenter, P.B. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 2004, 5, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Khair, L.; Guikema, J.E.; Linehan, E.K.; Ucher, A.J.; Leus, N.G.; Ogilvie, C.; Lou, Z.; Schrader, C.E.; Stavnezer, J. ATM increases activation-induced cytidine deaminase activity at downstream S regions during class-switch recombination. J. Immunol. 2014, 192, 4887–4896. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Wuerffel, R.; Wang, L.; Grigera, F.; Manis, J.; Selsing, E.; Perlot, T.; Alt, F.W.; Cogne, M.; Pinaud, E.; Kenter, A.L. S–S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity 2007, 27, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Rocha, P.P.; Raviram, R.; Fu, Y.; Kim, J.; Luo, V.M.; Aljoufi, A.; Swanzey, E.; Pasquarella, A.; Balestrini, A.; Miraldi, E.R.; et al. A Damage-Independent Role for 53BP1 that Impacts Break Order and Igh Architecture during Class Switch Recombination. Cell Rep. 2016, 16, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.; Wuerffel, R.; Achour, I.; Wang, L.; Carpenter, P.B.; Kenter, A.L. 53BP1 Contributes to Igh Locus Chromatin Topology during Class Switch Recombination. J. Immunol. 2017, 198, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Chaudhuri, J.; Alpert, C.; Dutt, S.; Ranganath, S.; Li, G.; Schrum, J.P.; Manis, J.P.; Alt, F.W. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature 2005, 438, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Vuong, B.Q.; Herrick-Reynolds, K.; Vaidyanathan, B.; Pucella, J.N.; Ucher, A.J.; Donghia, N.M.; Gu, X.; Nicolas, L.; Nowak, U.; Rahman, N.; et al. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat. Immunol. 2013, 14, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gostissa, M.; Hildebrand, D.G.; Becker, M.S.; Boboila, C.; Chiarle, R.; Lewis, S.; Alt, F.W. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv. Immunol. 2010, 106, 93–133. [Google Scholar] [PubMed]
- Neri, A.; Barriga, F.; Knowles, D.M.; Magrath, I.T.; Dalla-Favera, R. Different regions of the immunoglobulin heavy-chain locus are involved in chromosomal translocations in distinct pathogenetic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1988, 85, 2748–2752. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef] [PubMed]
- Di Noia, J.M.; Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.; Bransteitter, R.; Petruska, J.; Goodman, M.F. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003, 424, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, A.R.; Jankovic, M.; Eisenreich, T.; Difilippantonio, S.; Chen-Kiang, S.; Muramatsu, M.; Honjo, T.; Nussenzweig, A.; Nussenzweig, M.C. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 2004, 118, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Duke, J.L.; Richter, D.J.; Vinuesa, C.G.; Goodnow, C.C.; Kleinstein, S.H.; Schatz, D.G. Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008, 451, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.-J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.A.; Resch, W.; Jankovic, M.; Oliveira, T.; Yamane, A.; Nakahashi, H.; Di Virgilio, M.; Bothmer, A.; Nussenzweig, A.; Robbiani, D.F.; et al. Translocation-Capture Sequencing Reveals the Extent and Nature of Chromosomal Rearrangements in B Lymphocytes. Cell 2011, 147, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Hakim, O.; Resch, W.; Yamane, A.; Klein, I.; Kieffer-Kwon, K.R.; Jankovic, M.; Oliveira, T.; Bothmer, A.; Voss, T.C.; Ansarah-Sobrinho, C.; et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 2012, 484, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Klein, F.; Diskin, R.; Scheid, J.F.; Gaebler, C.; Mouquet, H.; Georgiev, I.S.; Pancera, M.; Zhou, T.; Incesu, R.B.; Fu, B.Z.; et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 2013, 153, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.-Y.; Mays, I.; Garman, L.; Helms, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Bemark, M.; Neuberger, M.S. By-products of immunoglobulin somatic hypermutation. Genes Chromosomes Cancer 2003, 38, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bowers, P.M.; Verdino, P.; Wang, Z.; da Silva Correia, J.; Chhoa, M.; Macondray, G.; Do, M.; Neben, T.Y.; Horlick, R.A.; Stanfield, R.L.; et al. Nucleotide insertions and deletions complement point mutations to massively expand the diversity created by somatic hypermutation of antibodies. J. Bio. Chem. 2014, 289, 33557–33567. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, K. Clonal selection and learning in the antibody system. Nature 1996, 381, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, A.D.; Shulman, Z.; Nussenzweig, M.C. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 2014, 509, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Peled, J.U.; Kuang, F.L.; Iglesias-Ussel, M.D.; Roa, S.; Kalis, S.L.; Goodman, M.F.; Scharff, M.D. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 2008, 26, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.; Maizels, N. Cell Cycle Regulates Nuclear Stability of AID and Determines the Cellular Response to AID. PLoS Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Krijger, P.H.; Langerak, P.; van den Berk, P.C.; Jacobs, H. Dependence of nucleotide substitutions on Ung2, Msh2, and PCNA-Ub during somatic hypermutation. J. Exp. Med. 2009, 206, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.G.; Langerak, P.; Tsaalbi-Shtylik, A.; van den Berk, P.; Jacobs, H.; de Wind, N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 2006, 203, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Linehan, E.K.; Thompson, M.R.; Habboub, G.; Ucher, A.J.; Kadungure, T.; Tsuchimoto, D.; Nakabeppu, Y.; Schrader, C.E. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation. Proc. Natl. Acad. Sci. USA 2014, 111, 9217–9222. [Google Scholar] [CrossRef] [PubMed]
- Poltoratsky, V.; Prasad, R.; Horton, J.K.; Wilson, S.H. Down-regulation of DNA polymerase beta accompanies somatic hypermutation in human BL2 cell lines. DNA Repair 2007, 6, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, J.M.; Roa, S.; Werling, U.; Liu, Y.; Genschel, J.; Hou, H., Jr.; Sellers, R.S.; Modrich, P.; Scharff, M.D.; Edelmann, W. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 13384–13389. [Google Scholar] [CrossRef] [PubMed]
- Methot, S.P.; Di Noia, J.M. Chapter Two-Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. In Advances in Immunology; Frederick, W.A., Ed.; Academic Press: Pittsburgh, PA, USA, 2017; Volume 133, pp. 37–87. [Google Scholar]
- Crouse, G.F. Non-canonical actions of mismatch repair. DNA Repair 2016, 38, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Chahwan, R.; Edelmann, W.; Scharff, M.D.; Roa, S. AIDing antibody diversity by error-prone mismatch repair. Semin. Immunol. 2012, 24, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M.S.; Harris, R.S.; Di Noia, J.; Petersen-Mahrt, S.K. Immunity through DNA deamination. Trends Biochem. Sci. 2003, 28, 305–312. [Google Scholar] [CrossRef]
- Pena-Diaz, J.; Bregenhorn, S.; Ghodgaonkar, M.; Follonier, C.; Artola-Boran, M.; Castor, D.; Lopes, M.; Sartori, A.A.; Jiricny, J. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. cell 2012, 47, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Langerak, P.; Nygren, A.O.; Krijger, P.H.; van den Berk, P.C.; Jacobs, H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007, 204, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Rada, C.; Di Noia, J.M.; Neuberger, M.S. Mismatch Recognition and Uracil Excision Provide Complementary Paths to Both Ig Switching and the A/T-Focused Phase of Somatic Mutation. Mol. Cell 2004, 16, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wiesendanger, M.; Kneitz, B.; Edelmann, W.; Scharff, M.D. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 2000, 191, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Martomo, S.A.; Yang, W.W.; Gearhart, P.J. A Role for Msh6 but Not Msh3 in Somatic Hypermutation and Class Switch Recombination. J. Exp. Med. 2004, 200, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Roa, S.; Li, Z.; Peled, J.U.; Zhao, C.; Edelmann, W.; Scharff, M.D. MSH2/MSH6 Complex Promotes Error-Free Repair of AID-Induced dU:G Mispairs as well as Error-Prone Hypermutation of A:T Sites. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Delbos, F.; Aoufouchi, S.; Faili, A.; Weill, J.C.; Reynaud, C.A. DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2007, 204, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.M.; Tanaka, A.; Bozek, G.; Nicolae, D.; Storb, U. Somatic hypermutation and class switch recombination in Msh6(-/-)Ung(-/-) double-knockout mice. J. Immunol. 2006, 177, 5386–5392. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, P.D.; Woo, C.J.; Wei, K.; Li, Z.; Martin, A.; Sack, S.Z.; Parris, T.; Edelmann, W.; Scharff, M.D. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 2004, 5, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Peters, A.; Baron, B.; Zhu, X.; Storb, U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 1998, 280, 1750–1752. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Schatz, D.G. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 2009, 30, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Bhagat, G.; Jankovic, M.; Compagno, M.; Smith, P.; Muramatsu, M.; Honjo, T.; Morse, H.C.; Nussenzweig, M.C.; Dalla-Favera, R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 2008, 40, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Yoshida, T.; Okada, S.; Hatano, M.; Miki, T.; Ishibashi, K.; Okabe, S.; Koseki, H.; Hirosawa, S.; Taniguchi, M.; et al. Disruption of the Bcl6 gene results in an impaired germinal center formation. J. Exp. Med. 1997, 186, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Dalla-Favera, R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol. Rev. 2012, 247, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Phan, R.T.; Dalla-Favera, R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004, 432, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Phan, R.T.; Saito, M.; Basso, K.; Niu, H.; Dalla-Favera, R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 2005, 6, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Polo, J.M.; Dierov, J.; Singer, M.; Kuo, T.; Greally, J.; Green, R.; Carroll, M.; Melnick, A. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat. Immunol. 2007, 8, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Polo, J.M.; Melnick, A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol. Dis. 2008, 41, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, H.S.; Galashevskaya, A.; Doseth, B.; Sousa, M.M.; Sarno, A.; Visnes, T.; Aas, P.A.; Liabakk, N.B.; Slupphaug, G.; Saetrom, P.; et al. AID expression in B-cell lymphomas causes accumulation of genomic uracil and a distinct AID mutational signature. DNA Repair 2015, 25, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2013. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lada, A.G.; Dhar, A.; Boissy, R.J.; Hirano, M.; Rubel, A.A.; Rogozin, I.B.; Pavlov, Y.I. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol. Direct 2012. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, Q.; Dose, M.; Pruett, N.; Kieffer-Kwon, K.R.; Resch, W.; Liang, G.; Tang, Z.; Mathe, E.; Benner, C.; et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 2014, 159, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Casellas, R.; Basu, U.; Yewdell, W.T.; Chaudhuri, J.; Robbiani, D.F.; Di Noia, J.M. Mutations, kataegis and translocations in B cells: Understanding AID promiscuous activity. Nat. Rev. Immunol. 2016, 16, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Kou, T.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Okazaki, I.M.; Ueda, Y.; Kodama, Y.; Haga, H.; Ikai, I.; Chiba, T. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int. J. Cancer 2007, 120, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Babbage, G.; Ottensmeier, C.H.; Blaydes, J.; Stevenson, F.K.; Sahota, S.S. Immunoglobulin heavy chain locus events and expression of activation-induced cytidine deaminase in epithelial breast cancer cell lines. Cancer Res. 2006, 66, 3996–4000. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Marusawa, H.; Kou, T.; Nakase, H.; Fujii, S.; Fujimori, T.; Kinoshita, K.; Honjo, T.; Chiba, T. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology 2008, 135, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Morisawa, T.; Marusawa, H.; Ueda, Y.; Iwai, A.; Okazaki, I.M.; Honjo, T.; Chiba, T. Organ-specific profiles of genetic changes in cancers caused by activation-induced cytidine deaminase expression. Int. J. Cancer 2008, 123, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Marusawa, H.; Chiba, T. Involvement of activation-induced cytidine deaminase in the development of colitis-associated colorectal cancers. J. Gastroenterol. 2011, 46, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, A.; Marusawa, H.; Minaki, Y.; Watanabe, T.; Nakase, H.; Kinoshita, K.; Tsujimoto, G.; Chiba, T. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene 2012, 31, 1733–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.-M.; Honjo, T.; Chiba, T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahjat, M.; Guikema, J.E.J. The Complex Interplay between DNA Injury and Repair in Enzymatically Induced Mutagenesis and DNA Damage in B Lymphocytes. Int. J. Mol. Sci. 2017, 18, 1876. https://doi.org/10.3390/ijms18091876
Bahjat M, Guikema JEJ. The Complex Interplay between DNA Injury and Repair in Enzymatically Induced Mutagenesis and DNA Damage in B Lymphocytes. International Journal of Molecular Sciences. 2017; 18(9):1876. https://doi.org/10.3390/ijms18091876
Chicago/Turabian StyleBahjat, Mahnoush, and Jeroen E. J. Guikema. 2017. "The Complex Interplay between DNA Injury and Repair in Enzymatically Induced Mutagenesis and DNA Damage in B Lymphocytes" International Journal of Molecular Sciences 18, no. 9: 1876. https://doi.org/10.3390/ijms18091876
APA StyleBahjat, M., & Guikema, J. E. J. (2017). The Complex Interplay between DNA Injury and Repair in Enzymatically Induced Mutagenesis and DNA Damage in B Lymphocytes. International Journal of Molecular Sciences, 18(9), 1876. https://doi.org/10.3390/ijms18091876