Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate



Abstract
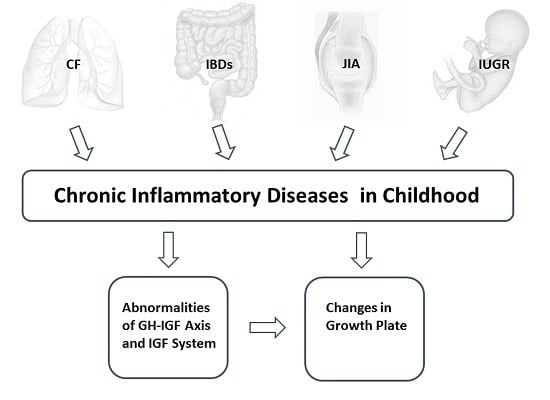
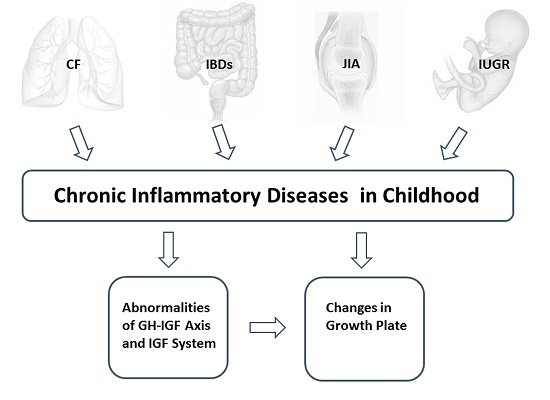
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Overview of Most Frequent Chronic Inflammatory Diseases with Impaired Growth
2.1. Cystic Fibrosis
2.2. Inflammatory Bowel Diseases
2.3. Juvenile Idiopathic Arthritis
2.4. Intrauterine Growth Restriction
3. Interactions between Pro-Inflammatory Cytokines and GH–IGF Axis, IGF System, and Bone
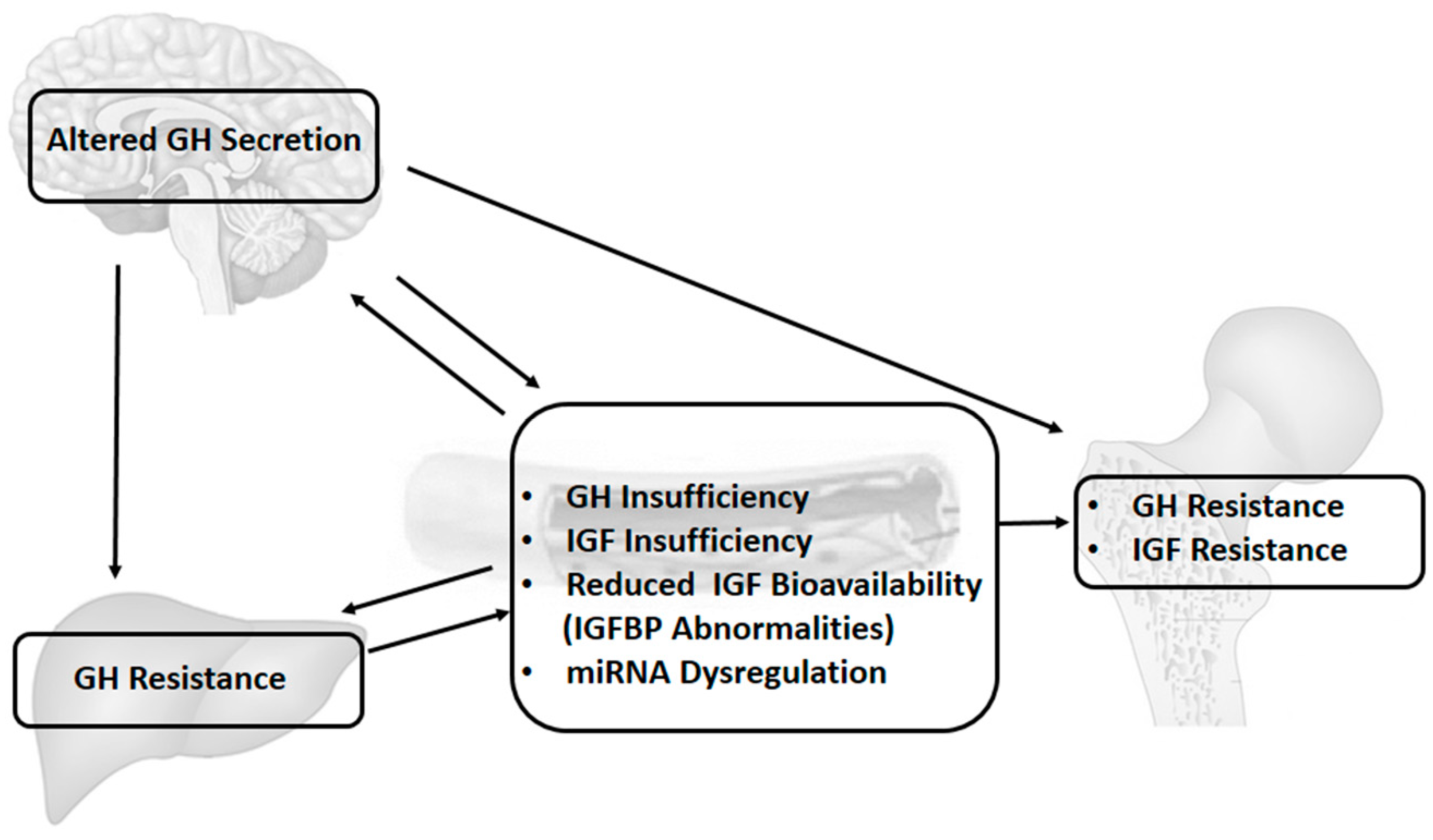
3.1. GH–IGF Axis and IGF System
3.2. Changes in the GH–IGF Axis and IGF System and Interactions with Pro-Inflammatory Cytokines
3.3. Inflammation and miRNAs
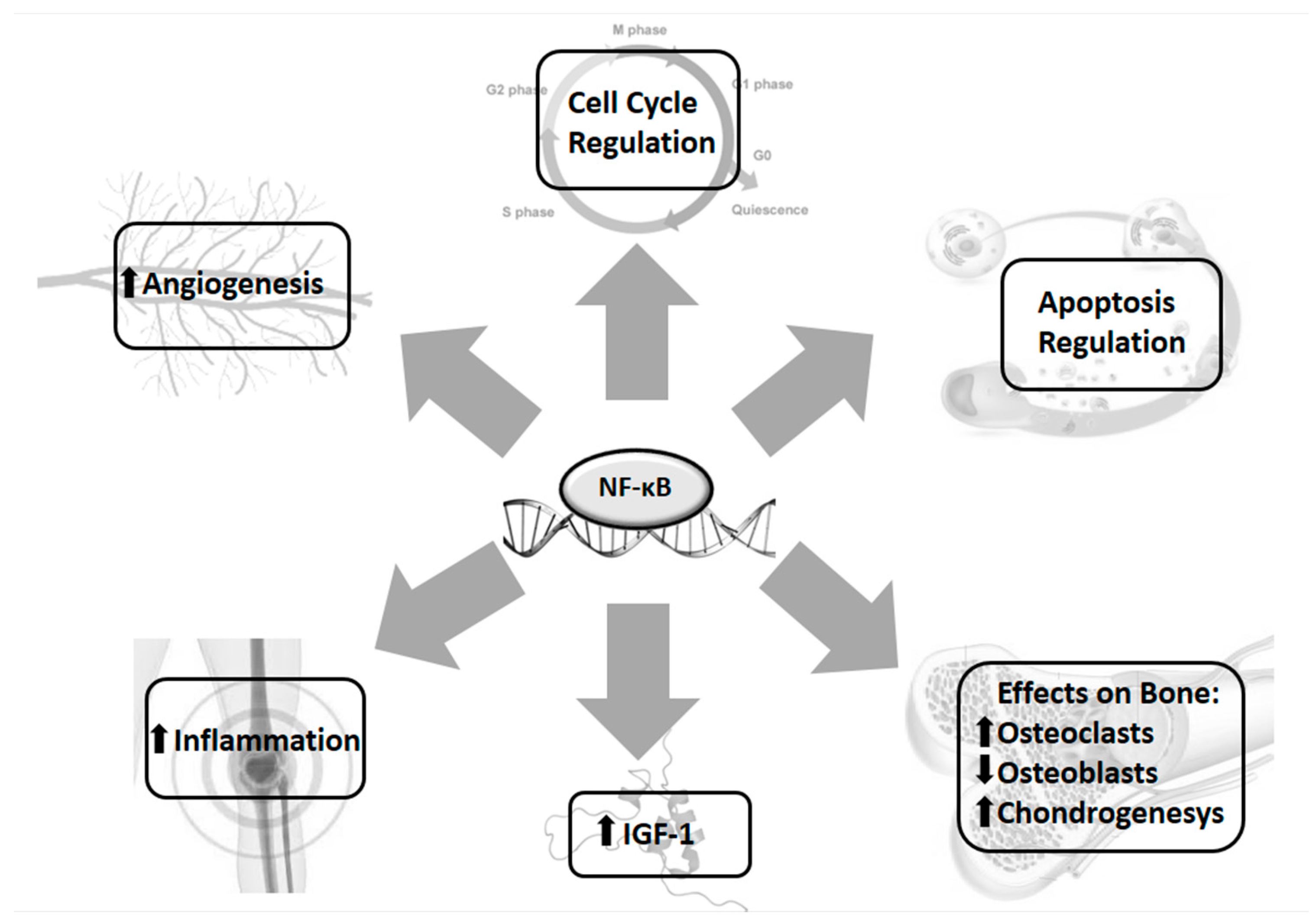
3.4. Bone Growth and Inflammation
4. Specific Changes in the GH–IGF Axis, IGF System, and Growth Plate in Individual Inflammatory Conditions
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the growth hormone-insulin like growth factor 1 axis in children with chronic inflammation: Current evidence, gaps in knowledge, and future directions. Endocr. Rev. 2015, 37, 62–110. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, I.R. Growth problems in children with IBD. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, B. Growth. Pediatr. Rev. 2011, 32, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N. Epigenetic influences that modulate infant growth, development, and disease. Antioxid. Redox Signal. 2012, 17, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Mullins, E.; Prior, T.; Roberts, I.; Kumar, S. Changes in the fetal and neonatal cytokine profile in pregnancies complicated by fetal growth restriction. Am. J. Reprod. Immunol. 2013, 69, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Street, M.E.; Seghini, P.; Fieni, S.; Ziveri, M.A.; Volta, C.; Martorana, D.; Viani, I.; Gramellini, D.; Bernasconi, S. Changes in interleukin-6 and IGF system and their relationships in placenta and cord blood in newborns with fetal growth restriction compared with controls. Eur. J. Endocrinol. 2006, 155, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Montanini, L.; Smerieri, A.; Gullì, M.; Cirillo, F.; Pisi, G.; Sartori, C.; Amarri, S.; Bernasconi, S.; Marmiroli, N.; Street, M.E. miR-146a, miR-155, miR-370, and miR-708 Are CFTR-Dependent, Predicted FOXO1 Regulators and Change at Onset of CFRDs. J. Clin. Endocrinol. Metab. 2016, 101, 4955–4963. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, F.; Kubik, Ł.; Oblak, A.; Lorè, N.I.; Cigana, C.; Lanzetta, R.; Parrilli, M.; Hamad, M.A.; De Soyza, A.; Silipo, A.; et al. Activation of human toll-like receptor 4 (TLR4) myeloid differentiation factor 2 (MD-2) by hypoacylated lipopolysaccharide from a clinical isolate of Burkholderia cenocepacia. J. Biol. Chem. 2015, 290, 21305–21319. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, D.; O’Connell, J. Cystic fibrosis related diabetes. Curr. Diabetes Rep. 2014, 4, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Schluchter, M.D.; Konstan, M.W.; Drumm, M.L.; Yankaskas, J.R.; Knowles, M.R. Classifying severity of cystic fibrosis lung disease using longitudinal pulmonary function data. Am. J. Respir. Crit. Care Med. 2006, 174, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Blackman, S.M.; Tangpricha, V. Endocrine disorders in cystic fibrosis. Pediatr. Clin. N. Am. 2016, 63, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.C.; Kosorok, M.R.; Sondel, S.A.; Chen, S.T.; FitzSimmons, S.C.; Green, C.G.; Shen, G.; Walker, S.; Farrell, P.M. Growth status in children with cystic fibrosis based on the National Cystic Fibrosis Patient Registry data: Evaluation of various criteria used to identify malnutrition. J. Pediatr. 1998, 132, 478–485. [Google Scholar] [CrossRef]
- Morison, S.; Dodge, J.A.; Cole, T.J.; Lewis, P.A.; Coles, E.C.; Geddes, D.; Russell, G.; Littlewood, J.M.; Scott, M.T. Height and weight in cystic fibrosis: A cross sectional study. Arch. Dis. Child. 1997, 77, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Bournez, M.; Bellis, G.; Huet, F. Growth during puberty in cystic fibrosis: A retrospective evaluation of a French cohort. Arch. Dis. Child. 2012, 97, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Aswani, N.; Taylor, C.J.; McGaw, J.; Pickering, M.; Rigby, A.S. Pubertal growth and development in cystic fibrosis: A retrospective review. Acta Paediatr. 2003, 92, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Vieni, G.; Faraci, S.; Collura, M.; Lombardo, M.; Traverso, G.; Cristadoro, S.; Termini, L.; Lucanto, M.C.; Furnari, M.L.; Trimarchi, G.; et al. Stunting is an independent predictor of mortality in patients with cystic fibrosis. Clin. Nutr. 2013, 32, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B. Inflammatory bowel disease: Epidemiology, pathogenesis, and therapeutic opportunities. Inflamm. Bowel Dis. 2006, 12, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.M.; Nguyen, P.; Smith, C.; MacMillan, J.H.; Sherman, P.M. Growth and clinical course of children with Crohn’s disease. Gut 1993, 34, 939–943. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, D.P.; Dawson, A.M. Crohn’s disease in childhood. Arch. Dis. Child. 1977, 52, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Gribetz, D.; Korelitz, B.I. Growth retardation in children with ulcerative colitis: The effect of medical and surgical therapy. Paediatrics 1975, 55, 459–467. [Google Scholar]
- Kirschner, B.S. Growth and development in chronic inflammatory bowel disease. Acta Paediatr. 1990, 79, 98–104. [Google Scholar] [CrossRef]
- Sullivan, D.B.; Cassidy, J.T.; Petty, R.E. Pathogenic implications of age of onset in juvenile rheumatoid arthritis. Arthritis Rheumatol. 1975, 18, 251–255. [Google Scholar] [CrossRef]
- Petty, R.E.; Southwood, T.R.; Baum, J.; Bhettay, E.; Glass, D.N.; Manners, P.; Maldonado-Cocco, J.; Suarez-Almazor, M.; Orozco-Alcala, J.; Prieur, A.M. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J. Rheumatol. 1998, 25, 1991–1994. [Google Scholar] [PubMed]
- Bruck, N.; Schnabel, A.; Hedrich, C.M. Current understanding of the pathophysiology of systemic juvenile idiopathic arthritis (sJIA) and target-directed therapeutic approaches. Clin. Immunol. 2015, 159, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, S.; Simon, D. Growth abnormalities in children and adolescents with juvenile idiopathic arthritis. Rheumatol. Int. 2014, 34, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- MacRae, V.E.; Ahmed, S.F.; Mushtaq, T.; Farquharson, C. IGF-I signalling in bone growth: Inhibitory actions of dexameth- asone and IL-1β. Growth Horm. IGF Res. 2007, 17, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packham, J.C.; Hall, M.A. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: Social function, relationships and sexual activity. Rheumatology 2002, 41, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.A.; Hoch, S.; Erlandson, D.; Partridge, R.; Jackson, J.M. The timing of menarche in juvenile rheumatoid arthritis. J. Adolesc. Health Care 1988, 9, 483–487. [Google Scholar] [CrossRef]
- Padeh, S.; Pinhas-Hamiel, O.; Zimmermann-Sloutskis, D.; Berkun, Y. Children with oligoarticular juvenile idiopathic arthritis are at considerable risk for growth retardation. J. Pediatr. 2011, 159, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Panera, N.; Agostoni, C.; Nobili, V. Intrauterine growth retardation and nonalcoholic Fatty liver disease in children. Int. J. Endocrinol. 2011, 2011, 269853. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine growth restriction: Antenatal and postnatal aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.M.; Horbar, J.D.; Badger, G.J.; Ohlsson, A.; Golan, A. Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction. Am. J. Obstet. Gynecol. 2000, 182, 198–206. [Google Scholar] [CrossRef]
- Street, M.E.; Volta, C.; Ziveri, M.A.; Viani, I.; Bernasconi, S. Markers of insulin sensitivity in placentas and cord serum of intrauterine growth-restricted newborns. Clin. Endocrinol. 2009, 71, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Martinez, C.; Maiorana, A.; Scirè, G.; Spadoni, G.L.; Boemi, S. Adiponectin levels are reduced in children born small for gestational age and are inversely related to postnatal catch-up growth. J. Clin. Endocrinol. Metab. 2004, 89, 1346–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.; Dziura, J.; Burgert, T.S.; Tamborlane, W.V.; Taksali, S.E.; Yeckel, C.W.; Allen, K.; Lopes, M.; Savoye, M.; Morrison, J.; et al. Obesity and the metabolic syndrome in children and adolescents. N. Engl. J. Med. 2004, 350, 2362–2374. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Maiorana, A.; Geremia, C.; Scirè, G.; Spadoni, G.L.; Germani, D. Blood glucose concentrations are reduced in children born small for gestational age (SGA), and thyroid-stimulating hormone levels are increased in SGA with blunted postnatal catch-up growth. J. Clin. Endocrinol. Metab. 2003, 88, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Möhlig, M.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003, 361, 226–228. [Google Scholar] [CrossRef]
- Veening, M.A.; Van Weissenbruch, M.M.; Delemarre-Van De Waal, H.A. Glucose tolerance, insulin sensitivity, and insulin secretion in children born small for gestational age. J. Clin. Endocrinol. Metab. 2002, 87, 4657–4661. [Google Scholar] [CrossRef] [PubMed]
- Hofman, P.L.; Cutfield, W.S.; Robinson, E.M.; Bergman, R.N.; Menon, R.K.; Sperling, M.A.; Gluckman, P.D. Insulin resistance in short children with intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1997, 82, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, J.; Albertsson-Wikland, K. Growth in full-term small-for-gestational-age infants: From birth to final height. Pediatr. Res. 1995, 38, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, O.G.; Eden, S.; Jansson, J.O. Mode of action of pituitary growth hormone on target cells. Annu. Rev. Physiol. 1985, 47, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef] [PubMed]
- Donahue, L.R.; Beamer, W.G. Growth hormone deficiency in ‘little’ mice results in aberrant body composition, reduced insulin-like growth factor-I and insulin-like growth factor-binding protein-3 (IGFBP-3), but does not affect IGFBP-2, -1 or -4. J. Endocrinol. 1993, 136, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.A.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (IGF-1) and type 1 IGF receptor (IGF1R). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Powell-Braxton, L.; Hollingshead, P.; Warburton, C.; Dowd, M.; Pitts-Meek, S.; Dalton, D.; Gillett, N.; Stewart, T.A. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993, 7, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Daughaday, W.H. From sulphation factor to IGF-I, 40 years of research on the regulation of cartilage growth. In Proceedings of the 4th International Symposium on Insulin-like Growth Factors, Tokyo, Japan, 21–24 October 1997; Takano, K., Hizuka, N., Takahashi, S.-I., Eds.; Elsevier: Tokyo, Japan, 1997. [Google Scholar]
- Carter-Su, C.; Schwartz, J.; Smit, L.S. Molecular mechanism of growth hormone action. Annu. Rev. Physiol. 1996, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Daughaday, W.H.; Hall, K.; Raben, M.S.; Salmon, W.D., Jr.; van Den Brande, J.L.; Van Wyk, J.J. Somatomedin: Proposed designation for sulphation factor. Nature 1972, 235, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bartold, P.M.; Zhang, C.Z.; Clarkson, R.W.; Young, W.G.; Waters, M.J. Growth hormone and insulin-like growth factor I induce bone morphogenetic proteins 2 and 4: A mediator role in bone and tooth formation? Endocrinology 1998, 139, 3855–3862. [Google Scholar] [CrossRef] [PubMed]
- Billestrup, N.; Nielsen, J.H. The stimulatory effect of growth hormone, prolactin, and placental lactogen on b-cell proliferation is not mediated by insulin-like growth factor-I. Endocrinology 1991, 129, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, O.G.; Lindahl, A.; Nilsson, A.; Isgaard, J. Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocr. Rev. 1987, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, A.; Nilsson, A.; Isaksson, O.G. Effects of growth hormone and insulin-like growth factor-I on colony formation of rabbit epiphyseal chondrocytes at different stages of maturation. J. Endocrinol. 1987, 115, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Schlechter, N.L.; Russell, S.M.; Spencer, E.M.; Nicoll, C.S. Evidence suggesting that the direct growth-promoting effect of growth hormone on cartilage in vivo is mediated by local production of somatomedin. Proc. Natl. Acad. Sci. USA 1986, 83, 7932–7934. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, O.G.; Jansson, J.O.; Gause, I.A. Growth hormone stimulates longitudinal bone growth directly. Science 1982, 216, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; LeRoith, D. Insulin-like growth factor I is essential for postnatal growth in response to growth hormone. Endocrinology 1999, 140, 5178–5184. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Insulin-like growth factor 1 (IGF-1): A growth hormone. Mol. Pathol. 2001, 54, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Blakesley, V.A.; Butler, A.A.; Koval, A.P.; Okubo, Y.; LeRoith, D. IGF-I receptor function: Transducing the IGF-1 signal into intracellular events. In The IGF System; Rosenfeld, R., Roberts, C., Jr., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 143–164. [Google Scholar]
- White, M.F.; Kahn, C.R. The insulin signalling system. J. Biol. Chem. 1994, 269, 1–4. [Google Scholar] [PubMed]
- Martin, J.L.; Baxter, R.C. IGF binding proteins and extracellular matrix. In The IGF System; Rosenfeld, R., Roberts, C., Jr., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 227–256. [Google Scholar]
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [PubMed]
- Holzenberger, M.; Leneuve, P.; Hamard, G.; Ducos, B.; Perin, L.; Binoux, M.; Le Bouc, Y. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology 2000, 141, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Alonzi, T.; Moretta, A.; Lazzaro, D.; Costa, P.; Poli, V.; Martini, A.; Ciliberto, G.; Fattori, E. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I-A model for stunted growth in children with chronic inflammation. J. Clin. Investig. 1997, 99, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Street, M.E.; Ziveri, M.A.; Spaggiari, C.; Viani, I.; Volta, C.; Grzincich, G.L.; Virdis, R.; Bernasconi, S. Inflammation is a modulator of the insulin-like growth factor (IGF)/IGF-binding protein system inducing reduced bioactivity of IGFs in cystic fibrosis. Eur. J. Endocrinol. 2006, 154, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Pass, C.; MacRae, V.E.; Huesa, C.; Faisal Ahmed, S.; Farquharson, C. SOCS2 is the critical regulator of GH action in murine growth plate chondrogenesis. J. Bone Miner Res. 2012, 27, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Farquharson, C. The effect of GH and IGF1 on linear growth and skeletal development and their modu- lation by SOCS proteins. J. Endocrinol. 2010, 206, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Held, M.A.; Menon, R.K.; Frank, S.J.; Parlow, A.F.; Arnold, D.L. Interleukin-6 inhibits hepatic growth hormone signaling via upregulation of Cis and Socs-3. Am. J. Physiol. 2003, 284, G646–G654. [Google Scholar] [CrossRef] [PubMed]
- Boisclair, Y.R.; Wang, J.; Shi, J.; Hurst, K.R.; Ooi, G. Role of the suppressor of cytokine signaling-3 in mediating the inhib- itory effects of interleukin-1β on the growth hormone-de- pendent transcription of the acid-labile subunit gene in liver cells. J. Biol. Chem. 2000, 275, 3841–3847. [Google Scholar] [CrossRef] [PubMed]
- Choukair, D.; Hügel, U.; Sander, A.; Uhlmann, L.; Tönshoff, B. Inhibition of IGF1-related intracellular signaling path- ways by proinflammatory cytokines in growth plate chondrocytes. Pediatr. Res. 2014, 76, 245–251. [Google Scholar] [CrossRef] [PubMed]
- MacRae, V.E.; Farquharson, C.; Ahmed, S.F. The restricted potential for recovery of growth plate chondrogenesis and longitudinal bone growth following exposure to pro-inflammatory cytokines. J. Endocrinol. 2006, 189, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Broussard, S.R.; McCusker, R.H.; Novakofski, J.E.; Strle, K.; Shen, W.H.; Johnson, R.W.; Dantzer, R.; Kelley, K.W. IL-1β impairs insulin-like growth factor i-induced differentiation and downstream activation signals of the insulin-like growth factor I receptor in myoblasts. J. Immunol. 2004, 172, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.; Gravallese, E.M. Impact of Inflammation on the Osteoblast in Rheumatic Diseases. Curr. Osteoporos. Rep. 2014, 12, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.P.; Lerner, U.H. The role of cytokines in inflammatory bone loss. Immunol. Investig. 2013, 42, 555–622. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Levenson, A.; Kharitonenkov, A.; De Luca, F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J. Biol. Chem. 2012, 287, 26060–26067. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Guo, N.J.; Tian, H.; Marohn, M.; Gearhart, S.; Bayless, T.M.; Brant, S.R.; Kwon, J.H. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; van Wengen, A.; Hoeve, M.A.; ten Dam, M.; van der Burg, M.; van Dongen, J.; van de Vosse, E.; van Tol, M.; Bredius, R.; Ottenhoff, T.H.; et al. The same IκBα mutation in two related individuals leads to completely different clinical syndromes. J. Exp. Med. 2004, 200, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-κB in osteoclast and B-cell development. Nature 1998, 392, 611–614. [Google Scholar]
- Kanegae, Y.; Tavares, A.T.; Izpisua Belmonte, J.C.; Verma, I.M. Role of Rel/NF-κB transcription factors during the outgrowth of the vertebrate limb. Genes Dev. 1997, 11, 3482–3496. [Google Scholar]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Ley, S.C. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Stoffel, M. MicroRNAs: A new class of regulatory genes affecting metabolism. Cell Metab. 2006, 4, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Iborra, M.; Bernuzzi, F.; Invernizzi, P.; Danese, S. MicroRNAs in autoimmunity and inflammatory bowel disease: Crucial regulators in immune response. Autoimmun. Rev. 2012, 11, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta 2009, 1792, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Good, L. Does everything now make (anti)sense? Biochem. Soc. Trans. 2006, 34, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ceppi, M.; Pereira, P.M.; Dunand-Sauthier, I.; Barras, E.; Reith, W.; Santos, M.A.; Pierre, P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocytederived dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- O’ Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Liston, A. MicroRNA in the immune system, microR NA as an immune system. Immunology 2009, 127, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signalling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Dunne, A.; O’Neill, L.A. Adaptor usage and Toll-like receptor signaling specificity. FEBS Lett. 2005, 579, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Kracht, M.; Saklatvala, J. Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine 2002, 20, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Tan, G.; Yang, C.H.; Fan, M.; Pfeffer, L.M.; Wu, Z.-H. DNA damage induces NF-κB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J. Biol. Chem. 2012, 287, 21783–21795. [Google Scholar] [CrossRef] [PubMed]
- Scisciani, C.; Vossio, S.; Guerrieri, F.; Schinzari, V.; De Laco, R.; D’Onorio De Meo, P.; Cervello, M.; Montalto, G.; Pollicino, T.; Raimondo, G.; et al. Transcriptional regulation of miR-224 upregulated in human HCCs by NFκB inflammatory pathways. J. Hepatol. 2012, 56, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, S.; Hu, T.; Liu, S.; He, Y.; Sun, S. Up-regulated microRNA-143 transcribed by nuclear factor κ B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 2009, 50, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Vencken, S.F.; Agrawal, R.; Gaughan, K.; Molloy, K.; Higgins, G.; McNally, P.; McElvaney, N.G.; Mall, M.A.; Greene, C.M. miR-17 overexpression in cystic fibrosis airway epithelial cells decreases interleukin-8 production. Eur. Respir. J. 2015, 46, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Cialfi, S.; Cimino, G.; De Biase, R.V.; Dominici, C.; Quattrucci, S.; Pizzuti, A. Elevated levels of miR-145 correlate with SMAD3 down-regulation in cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. MiR-126 is downregulated in cystic fibrosis airway epithelial cells and regulates TOM1 expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, S.; Yu, Q.; Yang, G.; Guo, J.; Li, M.; Zeng, Z.; He, Y.; Chen, B.; Chen, M. Circulating MicroRNA223 is a New Biomarker for Inflammatory Bowel Disease. Medicine 2016, 95, e2703. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Oikonomopoulos, A.; Mahurkar, S.; Touroutoglou, A.; Koukos, G.; Hommes, D.W.; Iliopoulos, D. Assessment of circulating microRNAs for the diagnosis and disease activity evaluation in patients with ulcerative colitis by using the nanostring technology. Inflamm. Bowel Dis. 2015, 21, 2533–2539. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus, C.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Nakamichi, I.; Esaki, M.; Asano, K.; Matsumoto, T.; Kitazono, T. Serum microRNA levels in patients with Crohn’s disease during induction therapy by infliximab. J. Gastroenterol. Hepatol. 2014, 29, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Plank, M.; Maltby, S.; Mattes, J.; Foster, P.S. Targeting translational control as a novel way to treat inflammatory disease: The emerging role of microRNAs. Clin. Exp. Allergy 2013, 43, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Duttagupta, R.; DiRienzo, S.; Jiang, R.; Bowers, J.; Gollub, J.; Kao, J.; Kearney, K.; Rudolph, D.; Dawany, N.B.; Showe, M.K.; et al. Genome-wide maps of circulating miRNA biomarkers for ulcerative colitis. PLoS ONE 2012, 7, e31241. [Google Scholar] [CrossRef] [PubMed]
- Paraskevi, A.; Theodoropoulos, G.; Papaconstantinou, I.; Mantzaris, G.; Nikiteas, N.; Gazouli, M. Circulating MicroRNA in inflammatory bowel disease. J. Crohn’s Colitis 2012, 6, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wu, F.; Xin, L.; Su, G.; He, F.; Yang, Y.; Sun, J.; Liu, Z. Differential plasma microRNAs expression in juvenile idiopathic arthritis. Mod. Rheumatol. 2016, 26, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Kawada, J.; Kawano, Y.; Torii, Y.; Kawabe, S.; Iwata, N.; Ito, Y. Serum microRNAs as potential biomarkers of juvenile idiopathic arthritis. Clin. Rheumatol. 2015, 34, 1705–1712. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Tsai, S.Q.; Hardison, N.E.; James, A.H.; Motsinger-Reif, A.A.; Thames, B.; Stone, E.A.; Deng, C.; Piedrahita, J.A. Differentially expressed microRNAs and affected biological pathways revealed by modulated modularity clustering (MMC) analysis of human preeclamptic and IUGR placentas. Placenta 2013, 34, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shen, Z.; Xu, Q.; Huang, X.; Chen, Q.; Li, D. Increased levels of microRNA-424 are associated with the pathogenesis of fetal growth restriction. Placenta 2013, 34, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, C.; Isgaard, J.; Tornell, J.; Nilsson, A.; Isaksson, O.G.P.; Lindahl, A. Endocrine regulation of longitudinal bone growth. Acta Paediatr. 1993, 391, 33–40. [Google Scholar] [CrossRef]
- Wit, J.M.; Boersma, B. Catch-up growth: Definition, mechanisms, and models. J. Pediatr. Endocrinol. Metab. 2002, 15, 1229–1241. [Google Scholar] [PubMed]
- Wit, J.M.; Camacho-Hübner, C. Endocrine regulation of longitudinal bone growth. Endocr. Dev. 2011, 21, 30–41. [Google Scholar] [PubMed]
- Sederquist, B.; Fernandez-Vojvodich, P.; Zaman, F.; Sävendahl, L. Recent research on the growth plate: Impact of inflammatory cytokines on longitudinal bone growth. J. Mol. Endocrinol. 2014, 53, T35–T44. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Cheng, C.M.; Kopchick, J.J.; Bondy, C.A. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J. Endocrinol. 2004, 180, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Shead, E.F.; Haworth, C.S.; Barker, H.; Bilton, D.; Compston, J.E. Osteoclast function, bone turnover and inflammatory cytokines during infective exacerbations of cystic fibrosis. J. Cyst. Fibros. 2010, 9, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, M.; Albanese, C.; Borrelli, O.; Civitelli, F.; Canitano, N.; Viola, F.; Passariello, R.; Cucchiara, S. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Romas, E.; Gillespie, M.T. Inflammation-induced bone loss: Can it be prevented? Rheum. Dis. Clin. N. Am. 2006, 32, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Iotsova, V.; Caamaño, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Hirata, S.; Shin, M.; Yamazaki, M.; Fukushima, H. Molecular mechanisms of BMP-induced bone formation: Cross-talk between BMP and NF-κB signaling pathways in osteoblastogenesis. Jpn. Dent. Sci. Rev. 2010, 46, 33–42. [Google Scholar] [CrossRef]
- Gilbert, L.C.; Rubin, J.; Nanes, M.S. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am. J. Physiol. Endocrinol. Metab. 2005, 13, 720–730. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F. Role of Nuclear Factor κ B (NF-κB) in Growth Plate Chondrogenesis. Pediatr. Endocrinol. Rev. 2016, 13, 720–730. [Google Scholar] [PubMed]
- Wu, S.; Flint, J.K.; Rezvani, G.; De Luca, F. Nuclear factor-κB p65 facilitates longitudinal bone growth by inducing growth plate chondrocyte proliferation and differentiation and by preventing apoptosis. J. Biol. Chem. 2007, 282, 33698–33706. [Google Scholar] [CrossRef] [PubMed]
- Crisostomo, P.R.; Wang, Y.; Markel, T.A.; Wang, M.; Lahm, T.; Meldrum, D.R. Human mesenchymal stem cells stimulated by TNF-α, LPS, or hypoxia produce growth factors by an NF-κB- but not JNK-dependent mechanism. Am. J. Physiol. Cell Physiol. 2008, 294, C675–C682. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Ko, J.Y.; Yeh, D.W.; Ke, H.C.; Wu, H.L. Modulation of dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation, and bone mass loss. Endocrinology 2008, 149, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Marcovecchio, M.L.; Mohn, A.; Chiarelli, F. Inflammatory cytokines and growth in childhood. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Eijken, M.; Koedam, M.; van Driel, M.; Buurman, C.J.; Pols, H.A.; van Leeuwen, J.P. The essential role of glucocorticoids for proper human osteoblast differentiation and matrix mineralization. Mol. Cell. Endocrinol. 2006, 248, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Burnham, J.M.; Shults, J.; Petit, M.A.; Semeao, E.; Beck, T.J.; Zemel, B.S.; Leonard, M.B. Alterations in proximal femur geometry in children treated withglucocorticoids for Crohn disease or nephrotic syndrome: Impact of the underlying disease. J. Bone Miner. Res. 2007, 22, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.B.; Feldman, H.I.; Shults, J.; Zemel, B.S.; Foster, B.J.; Stallings, V.A. Long-term, high-dose glucocorticoids and bone mineral content in childhood glucocorticoid-sensitive nephrotic syndrome. N. Engl. J. Med. 2004, 351, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Ciro, D.; Padoan, R.; Blau, H.; Marostica, A.; Fuoti, M.; Volpi, S.; Pilotta, A.; Meyerovitch, J.; Sher, D.; Assael, B.M. Growth retardation and reduced growth hormone secretion in cystic fibrosis. Clinical observations from three CF centers. J. Cyst. Fibros. 2013, 12, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Ripa, P.; Robertson, I.; Cowley, D.; Harris, M.; Masters, I.B.; Cotterill, A.M. The relationship between insulin secretion, the insulin-like growth factor axis and growth in children with cystic fibrosis. Clin. Endocrinol. 2002, 56, 383–389. [Google Scholar] [CrossRef]
- Laursen, T.; Jørgensen, J.O.; Jakobsen, G.; Hansen, B.L.; Christiansen, J.S. Continuous infusion versus daily injections of growth hormone (GH) for 4 weeks in GH-deficient patients. J. Clin. Endocrinol. Metab. 1995, 80, 2410–2418. [Google Scholar] [PubMed]
- Liang, H.; Yang, L.; Ma, T.; Zhao, Y. Functional expression of cystic fibrosis transmembrane conductance regulator in mouse chondrocytes. Clin. Exp. Pharmacol. Physiol. 2010, 37, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Diwakar, A.; Adam, R.J.; Michalski, A.S.; Tamegnon, M.M.; Fischer, A.J.; Launspach, J.L.; Horan, R.A.; Kao, S.C.; Chaloner, K.; Meyerholz, D.K.; et al. Sonographic evidence of abnormal tracheal cartilage ring structure in cystic fibrosis. Laryngoscope 2015, 125, 2398–2404. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, E.; Le Rouzic, P.; Bernaudin, J.F.; Cottart, C.H.; Vandebrouck, C.; Crié, A.; Leal, T.; Clement, A.; Bonora, M. Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulatordeficient mice. J. Physiol. 2008, 586, 3231–3243. [Google Scholar] [CrossRef] [PubMed]
- Ketelslegers, J.M.; Maiter, D.; Maes, M.; Underwood, L.E.; Thissen, J.P. Nutritional regulation of the growth hormone and insulin-like growth factor-binding proteins. Horm. Res. 1996, 45, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, A.B.; Azooz, O.; El-Haj, T.; Poole, S.; Farthing, M.J. Growth failure occurs through a decrease in insulin-like growth factor 1 which is independent of undernutrition in a rat model of colitis. Gut 2000, 46, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, A. Fundamental mechanisms of growth failure in inflammatory bowel disease. Horm. Res. 2002, 58, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Irwin, R.; Raehtz, S.; Parameswaran, N.; McCabe, L.R. Intestinal inflammation without weight loss decreases bone density and growth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1149–R1157. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Meazza, C.; Oliveri, M.; Pignatti, P.; Vivarelli, M.; Alonzi, T.; Fattori, E.; Garrone, S.; Barreca, A.; Martini, A. Effect of IL-6 on IGF binding protein-3: A study in IL-6 trans- genic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology 2001, 142, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Templ, E.; Koeller, M.; Riedl, M.; Wagner, O.; Graninger, W.; Luger, A. Anterior pituitary function in patients with newly diagnosed rheumatoid arthritis. Br. J. Rheumatol. 1996, 35, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Bergad, P.L.; Schwarzenberg, S.J.; Humbert, J.T.; Morrison, M.; Amar- asinghe, S.; Towle, H.C.; Berry, S.A. Inhibition of growth hor- mone action in models of inflammation. Am. J. Physiol. Cell Physiol. 2000, 279, C1906–C1917. [Google Scholar] [PubMed]
- Von Laue, S.; Ross, R.J. Inflammatory cytokines and acquired growth hormone resistance. Growth Horm. IGF Res. 2000, 10, S9–S14. [Google Scholar] [CrossRef]
- Bozzola, E.; Pagani, S.; Meazza, C.; Cortis, E.; Lisini, D.; Laarej, K.; Bozzola, M. Changes in growth hormone receptor gene expression during therapy in children with juvenile idiopathic arthritis. Horm. Res. Paediatr. 2012, 77, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Calderon, A.; Ibáñez de Caceres, I.; Soto, L.; Priego, T.; Martin, A.I.; Villanua, M.A. The decrease in hepatic IGF-I gene expression in arthritic rats is not associated with modifications in hepatic GH receptor mRNA. Eur. J. Endocrinol. 2001, 144, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.; Martín, A.I.; Villanúa, M.A.; López-Calderón, A. Experimental arthritis inhibits the insulin-like growth factor-I axis and induces muscle wasting through cyclooxygenase-2 activation. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1656–E1665. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Calderón, A.; Soto, L.; Martín, A.I. Chronic inflammation inhibits GH secretion and alters the serum insulin-like growth factor system in rats. Life Sci. 1999, 65, 2049–2060. [Google Scholar] [CrossRef]
- Wong, S.C.; MacRae, V.E.; Gracie, J.A.; McInnes, I.B.; Galea, P.; Gardner-Medwin, J.; Ahmed, S.F. Inflammatory cytokines in juvenile idiopathic arthritis: Effects on physical growth and the insulin-like-growth factor axis. Growth Horm. IGF Res. 2008, 18, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, C.; Pap, T.; Buckley, C.D.; Naylor, A.J. The role of stromal cells in inflammatory bone loss. Clin. Exp. Immunol. 2017, 189, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, S.; Marcovecchio, M.L.; Breda, L.; Chiarelli, F. Growth in juvenile idiopathic arthritis: The role of inflammation. Clin. Exp. Immunol. 2011, 29, 104–110. [Google Scholar]
- Bowman, C.J.; Streck, R.D.; Chapin, R.E. Maternal-placental insulin-like growth factor (IGF) signaling and its importance to normal embryo-fetal development. Birth Defects Res. B 2010, 89, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Kanaka-Gantenbein, C.; Mastorakos, G.; Chrousos, G.P. Endocrine-related causes and consequences of intrauterine growth retardation. Ann. N. Y. Acad. Sci. 2003, 997, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Smerieri, A.; Petraroli, M.; Ziveri, M.A.; Volta, C.; Bernasconi, S.; Street, M.E. Effects of cord serum insulin, IGF-II, IGFBP-2, IL-6 and cortisol concentrations on human birth weight and length: Pilot study. PLoS ONE 2011, 6, e29562. [Google Scholar] [CrossRef] [PubMed]
- Toure, D.M.; Baccaglini, L.; Opoku, S.T.; Barnes-Josiah, D.; Cox, R.; Hartman, T.; Klinkebiel, D. Epigenetic dysregulation of Insulin-like growth factor (IGF)-related genes and adverse pregnancy outcomes: A systematic review. J. Matern. Fetal. Neonatal. Med. 2016, 29, 3542–3552. [Google Scholar] [CrossRef] [PubMed]
- Völkl, T.M.; Schwöbel, K.; Simm, D.; Beier, C.; Rohrer, T.R.; Dörr, H.G. Spontaneous growth hormone secretion and IGF1:IGFBP3 molar ratios in children born small for gestational age (SGA). Growth Horm. IGF Res. 2004, 14, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Boguszewski, M.; Rosberg, S.; Albertsson-Wikland, K. Spontaneous 24-hour growth hormone profiles in prepubertal small for gestational age children. J. Clin. Endocrinol. Metab. 1995, 80, 2599–2606. [Google Scholar] [PubMed]
- De Waal, W.J.; Hokken-Koelega, A.C.; Stijnen, T.; de Muinck Keizer-Schrama, S.M.; Drop, S.L. Endogenous and stimulated GH secretion, urinary GH excretion, and plasma IGF-I and IGF-II levels in prepubertal children with short stature after intrauterine growth retardation. Clin. Endocrinol. 1994, 41, 621–630. [Google Scholar] [CrossRef]
- Philip, A.G. Fetal growth retardation: Femurs, fontanels, and follow-up. Pediatrics 1978, 62, 446–453. [Google Scholar] [PubMed]
- Wilson, M.G.; Meyers, H.I.; Peters, A.H. Postnatal bone growth of infants with fetal growth retardation. Pediatrics 1967, 40, 213–223. [Google Scholar] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirillo, F.; Lazzeroni, P.; Sartori, C.; Street, M.E. Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. Int. J. Mol. Sci. 2017, 18, 1878. https://doi.org/10.3390/ijms18091878
Cirillo F, Lazzeroni P, Sartori C, Street ME. Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. International Journal of Molecular Sciences. 2017; 18(9):1878. https://doi.org/10.3390/ijms18091878
Chicago/Turabian StyleCirillo, Francesca, Pietro Lazzeroni, Chiara Sartori, and Maria Elisabeth Street. 2017. "Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate" International Journal of Molecular Sciences 18, no. 9: 1878. https://doi.org/10.3390/ijms18091878
APA StyleCirillo, F., Lazzeroni, P., Sartori, C., & Street, M. E. (2017). Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. International Journal of Molecular Sciences, 18(9), 1878. https://doi.org/10.3390/ijms18091878