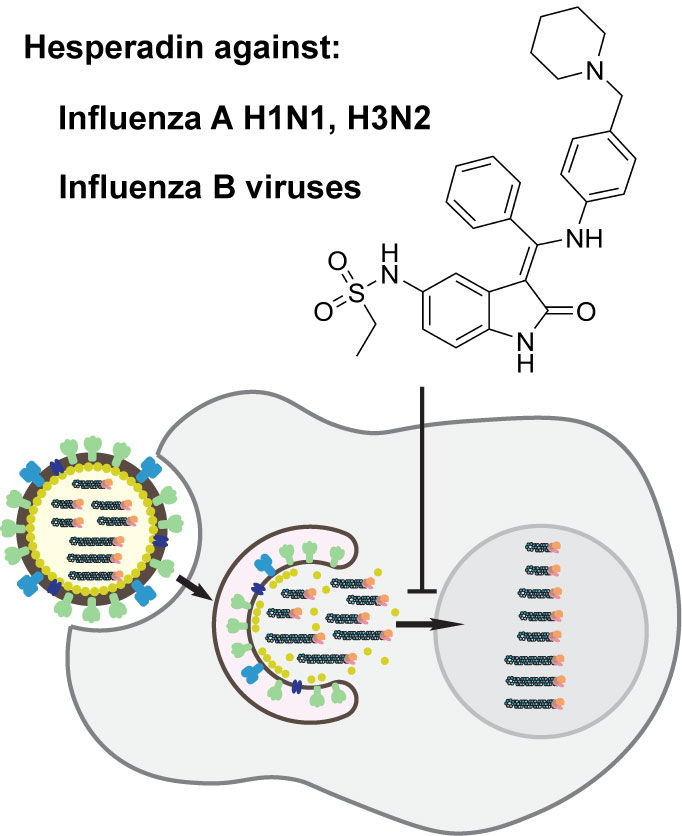
Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral


Abstract
:
1. Introduction
2. Results
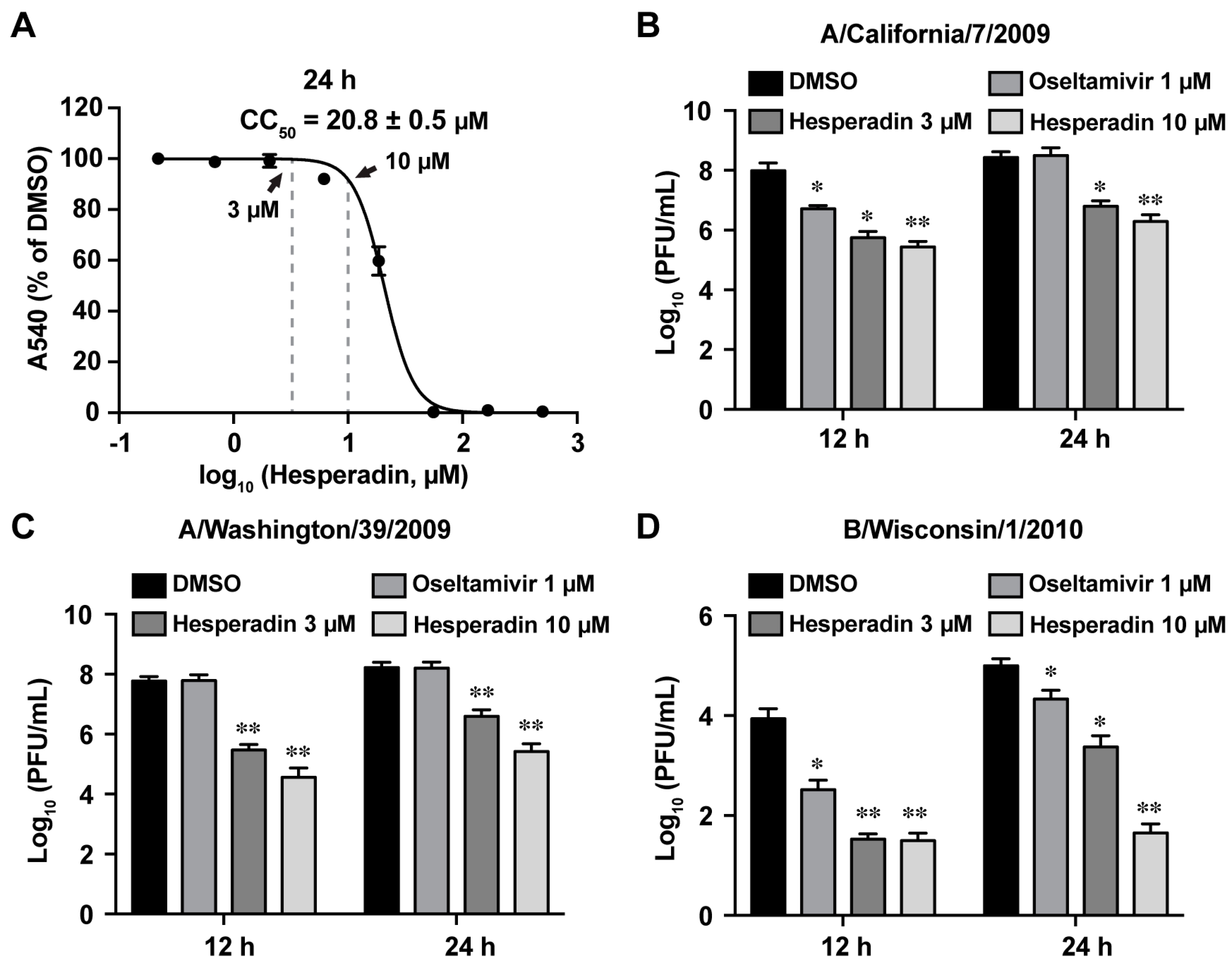
2.1. Hesperadin Inhibits Multiple Human Clinic Isolates of Influenza A and B Viruses
2.2. Hesperadin Inhibits Viral Replication in Cells Infected with a High Multiplicity of Infection (MOI)
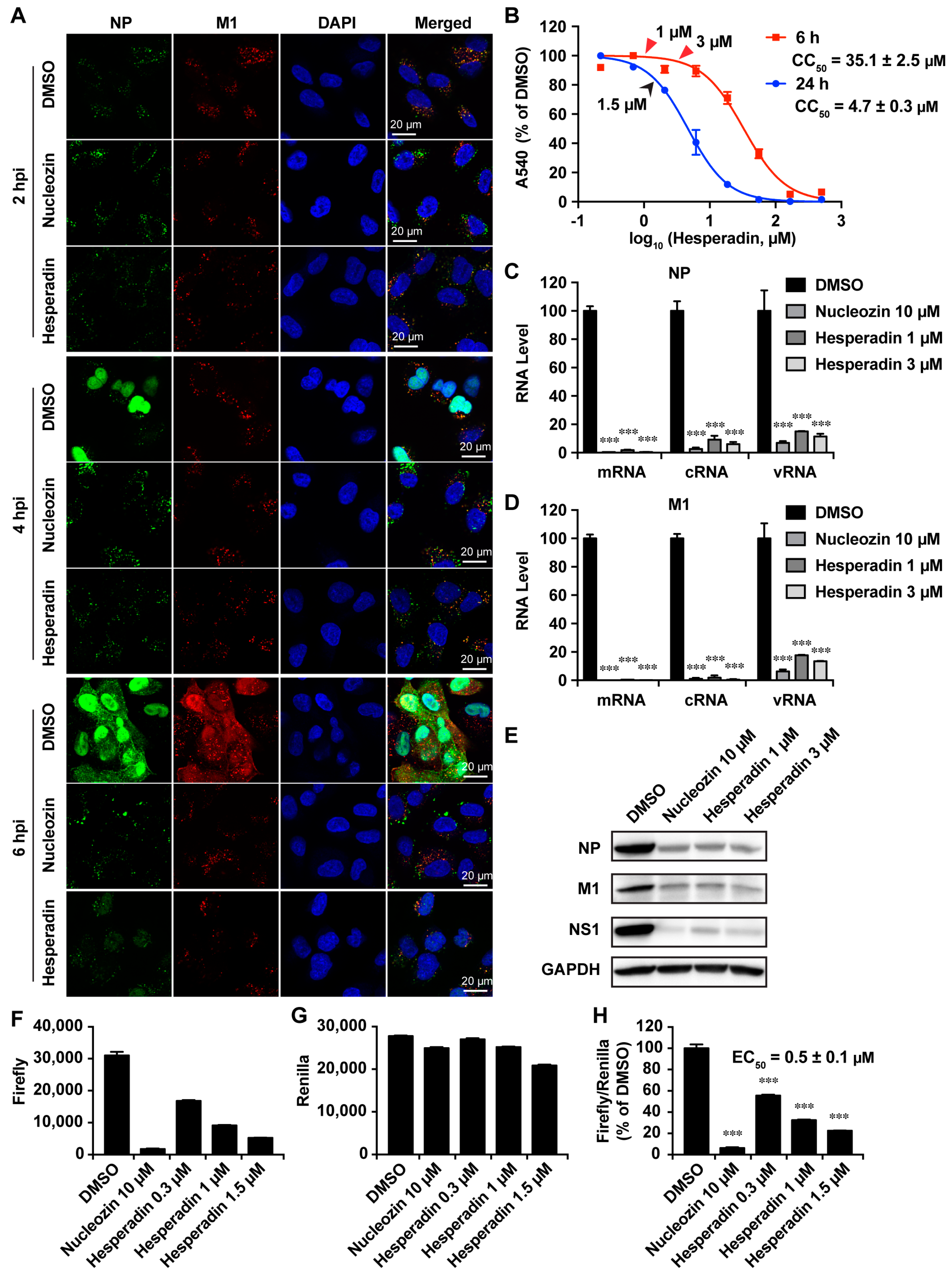
2.3. Hesperadin Inhibits the Early Stage of Viral Replication Post Viral Entry
2.4. Hesperadin Inhibits Viral RNA Transcription, Translation and Viral Protein Synthesis
2.5. Hesperadin Shows Synergistic Antiviral Activity with Oseltamivir in Combination Therapy
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Viruses, and Viral Infection
4.2. Plaque Assay
4.3. Cytotoxicity Assay and Cytopathic Effect (CPE) Assay
4.4. Time-of-Addition Experiment
4.5. Influenza Virus Minigenome Assay
4.6. RNA Extraction and Real-Time PCR
4.7. Western Blotting
4.8. Immunostaining
4.9. Assessment of Combination Treatment of Hesperadin with Oseltamivir In Vitro
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HA | Hemagglutinin |
| NA | Neuraminidase |
| MOI | Multiplicity of infection |
| CPE | Cytopathic effect |
| vRNP | Viral ribonucleoprotein |
| RT-qPCR | Real-time quantitative polymerase chain reaction |
| FICI | Fractional inhibitory concentration index |
| MDCK | Madin–Darby canine kidney |
References
- Webster, R.G.; Monto, A.S.; Braciale, T.J.; Lamb, R.A. Textbook of Influenza; Wiley: New York, NY, USA, 2013. [Google Scholar]
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The Viruses and Their Replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1647–1690. [Google Scholar]
- Krammer, F.; Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef] [PubMed]
- People at High Risk of Developing Flu–Related Complications. Available online: https://www.cdc.gov/flu/about/disease/high_risk.htm (accessed on 15 July 2017).
- Lai, S.; Qin, Y.; Cowling, B.J.; Ren, X.; Wardrop, N.A.; Gilbert, M.; Tsang, T.K.; Wu, P.; Feng, L.; Jiang, H.; et al. Global epidemiology of avian influenza A H5N1 virus infection in humans, 1997–2015: A systematic review of individual case data. Lancet Infect. Dis. 2016, 16, e108–e118. [Google Scholar] [CrossRef]
- Zhu, H.; Lam, T.T.; Smith, D.K.; Guan, Y. Emergence and development of H7N9 influenza viruses in China. Curr. Opin. Virol. 2016, 16, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, F.; Ma, C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers 2015, 104, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Loregian, A.; Mercorelli, B.; Nannetti, G.; Compagnin, C.; Palu, G. Antiviral strategies against influenza virus: Towards new therapeutic approaches. Cell. Mol. Life Sci. 2014, 71, 3659–3683. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-resistant influenza a viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C. The epidemiology and spread of drug resistant human influenza viruses. Curr. Opin. Virol. 2014, 8, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Sheu, T.; Fry, A.; Garten, R.; Deyde, V.; Shwe, T.; Bullion, L.; Peebles, P.; Li, Y.; Klimov, A.; Gubareva, L. Dual Resistance to Adamantanes and Oseltamivir Among Seasonal Influenza A(H1N1) Viruses: 2008–2010. J. Infect. Dis. 2011, 203, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Burnham, A.J.; Baranovich, T.; Govorkova, E.A. Neuraminidase inhibitors for influenza B virus infection: Efficacy and resistance. Antivir. Res. 2013, 100, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Santesso, N.; Mustafa, R.; Brozek, J.; Chen, Y.L.; Hopkins, J.P.; Cheung, A.; Hovhannisyan, G.; Ivanova, L.; Flottorp, S.A.; et al. Antivirals for treatment of influenza: A systematic review and meta-analysis of observational studies. Ann. Intern. Med. 2012, 156, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Koszalka, P.; Tilmanis, D.; Hurt, A.C. Influenza antivirals currently in late-phase clinical trial. Influenza Other Respir. Viruses 2017, 11, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.L. The host interactome of influenza virus presents new potential targets for antiviral drugs. Rev. Med. Virol. 2011, 21, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Sakurai, A.; Watanabe, T.; Sorensen, E.; Nidom, C.A.; Newton, M.A.; Ahlquist, P.; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, B.; Bamba, D.; Weng, K.; Ung, H.; Chang, S.; Van Dyke, J.; Goldblatt, M.; Duan, R.; Kinch, M.S.; Li, W.B. The use of Random Homozygous Gene Perturbation to identify novel host-oriented targets for influenza. Virology 2009, 387, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kawakami, E.; Shoemaker, J.E.; Lopes, T.J.; Matsuoka, Y.; Tomita, Y.; Kozuka-Hata, H.; Gorai, T.; Kuwahara, T.; Takeda, E.; et al. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe 2014, 16, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Hauf, S.; Cole, R.W.; LaTerra, S.; Zimmer, C.; Schnapp, G.; Walter, R.; Heckel, A.; van Meel, J.; Rieder, C.L.; Peters, J.M. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003, 161, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, J.; Wang, J. Pharmacological Characterization of the Spectrum of Antiviral Activity and Genetic Barrier to Drug Resistance of M2-S31N Channel Blockers. Mol. Pharmacol. 2016, 90, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Deyde, V.M.; Sheu, T.G.; Trujillo, A.A.; Okomo-Adhiambo, M.; Garten, R.; Klimov, A.I.; Gubareva, L.V. Detection of molecular markers of drug resistance in 2009 pandemic influenza A (H1N1) viruses by pyrosequencing. Antimicrob. Agents Chemother. 2010, 54, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Sakai-Tagawa, Y.; Kiso, M.; Goto, H.; Kawakami, C.; Mitamura, K.; Sugaya, N.; Suzuki, Y.; Kawaoka, Y. Enhanced Expression of an α2,6-Linked Sialic Acid on MDCK Cells Improves Isolation of Human Influenza Viruses and Evaluation of Their Sensitivity to a Neuraminidase Inhibitor. J. Clin. Microbiol. 2005, 43, 4139–4146. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Musharrafieh, R.; Ma, C.; Zhang, J.; Smee, D.F.; DeGrado, W.F.; Wang, J. An M2-V27A channel blocker demonstrates potent in vitro and in vivo antiviral activities against amantadine-sensitive and -resistant influenza A viruses. Antivir. Res. 2017, 140, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, F.; Musharrafieh, R.G.; Wang, J. Discovery of cyclosporine A and its analogs as broad-spectrum anti-influenza drugs with a high in vitro genetic barrier of drug resistance. Antivir. Res. 2016, 133, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sneyd, H.; Dekant, R.; Wang, J. Influenza A Virus Nucleoprotein: A Highly Conserved Multi-Functional Viral Protein As A Hot Antiviral Drug Target. Curr. Top. Med. Chem. 2017, 17, 2271–2285. [Google Scholar] [CrossRef] [PubMed]
- Beyleveld, G.; White, K.M.; Ayllon, J.; Shaw, M.L. New-generation screening assays for the detection of anti-influenza compounds targeting viral and host functions. Antivir. Res. 2013, 100, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Au, J.L.; Wientjes, M.G. Comparison of methods for evaluating drug-drug interaction. Front. Biosci. Elite Ed. 2010, 2, 241–249. [Google Scholar] [PubMed]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, M.C. What is synergy? Pharmacol. Rev. 1989, 41, 93–141. [Google Scholar] [PubMed]
- Lee, S.M.; Yen, H.L. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antivir. Res. 2012, 96, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Pohl, M.O.; Zhou, Y.Y.; Rodriguez-Frandsen, A.; Wang, G.J.; Stein, D.A.; Moulton, H.M.; DeJesus, P.; Che, J.W.; Mulder, L.C.F.; et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 2015, 18, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; e Sousa, C.R. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Roncal, N.E.; Lee, P.J.; Leed, S.E.; Erath, J.; Rodriguez, A.; Sciotti, R.J.; Pollastri, M.P. Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. Med. Chem. Commun. 2014, 5, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ma, C.; DeGrado, W.F.; Wang, J. Discovery of Highly Potent Inhibitors Targeting the Predominant Drug-Resistant S31N Mutant of the Influenza A Virus M2 Proton Channel. J. Med. Chem. 2016, 59, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Huggins, J.W.; Robins, R.K.; Canonico, P.G. Synergistic antiviral effects of ribavirin and the C-nucleoside analogs tiazofurin and selenazofurin against togaviruses, bunyaviruses, and arenaviruses. Antimicrob. Agents Chemother. 1984, 26, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Influenza Strains | Drug Sensitivity | EC50 (µM) a | SI b |
|---|---|---|---|
| A/Texas/4/2009 (H1N1) | 1.07 ± 0.11 | 19.9 | |
| A/North Carolina/39/2009 (H1N1) | 0.82 ± 0.02 | 26 | |
| A/Washington/29/2009 (H1N1) | Amantadine Resistant | 0.37 ± 0.12 | 57.6 |
| A/California/02/2014 (H3N2) | Oseltamivir Resistant | 1.80 ± 0.42 | 11.8 |
| A/Texas/12/2007 (H3N2) | 2.21 ± 0.23 | 9.7 | |
| A/WSN/1933 (H1N1) | Amantadine Resistant | 0.22 ± 0.04 | 56.1 |
| A/California/07/2009 (H1N1) | Oseltamivir Sensitive | 0.28 ± 0.03 | 76.1 |
| B/Wisconsin/1/2010 (Yamagata) | Amantadine Resistant | 0.73 ± 0.05 | 29.2 |
| B/Brisbane/60/2008 (Victoria) | Oseltamivir Sensitive | 0.52 ± 0.05 | 41 |
| Combination Ratio (EC50) | EC50 in Combination | EC50 Alone | EC50 Equivalent a | FICI b | |||
|---|---|---|---|---|---|---|---|
| Hesperadin: Oseltamivir | Hesperadin (μM) | Oseltaimivir (nM) | Hesperadin (μM) | Oseltaimivir (nM) | Hesperadin | Oseltaimivir | |
| 10:1 | 0.48 ± 0.15 | 1.1 ± 0.25 | 0.238 | 0.106 | 0.34 | ||
| 5:1 | 0.56 ± 0.05 | 2.26 ± 0.20 | 0.277 | 0.217 | 0.49 | ||
| 1:1 | 0.17 ± 0.20 | 3.44 ± 1.01 | 2.02 ± 0.18 | 10.10 ± 3.11 | 0.084 | 0.331 | 0.41 |
| 1:5 | 0.037 ± 0.006 | 3.73 ± 0.62 | 0.018 | 0.359 | 0.38 | ||
| 1:10 | 0.034 ± 0.052 | 6.28 ± 1.17 | 0.017 | 0.604 | 0.62 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Zhang, J.; Musharrafieh, R.; Hau, R.; Ma, C.; Wang, J. Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral. Int. J. Mol. Sci. 2017, 18, 1929. https://doi.org/10.3390/ijms18091929
Hu Y, Zhang J, Musharrafieh R, Hau R, Ma C, Wang J. Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral. International Journal of Molecular Sciences. 2017; 18(9):1929. https://doi.org/10.3390/ijms18091929
Chicago/Turabian StyleHu, Yanmei, Jiantao Zhang, Rami Musharrafieh, Raymond Hau, Chunlong Ma, and Jun Wang. 2017. "Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral" International Journal of Molecular Sciences 18, no. 9: 1929. https://doi.org/10.3390/ijms18091929
APA StyleHu, Y., Zhang, J., Musharrafieh, R., Hau, R., Ma, C., & Wang, J. (2017). Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral. International Journal of Molecular Sciences, 18(9), 1929. https://doi.org/10.3390/ijms18091929