Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species

 ,
, 

Abstract
:1. Introduction
2. Mitochondria and Epilepsy—Adenosine Triphosphate (ATP), Ca2+ and Cell Death
2.1. ATP during Seizures and Epilepsy
2.2. Mitochondria and Ca2+ Buffering in Seizures and Epilepsy
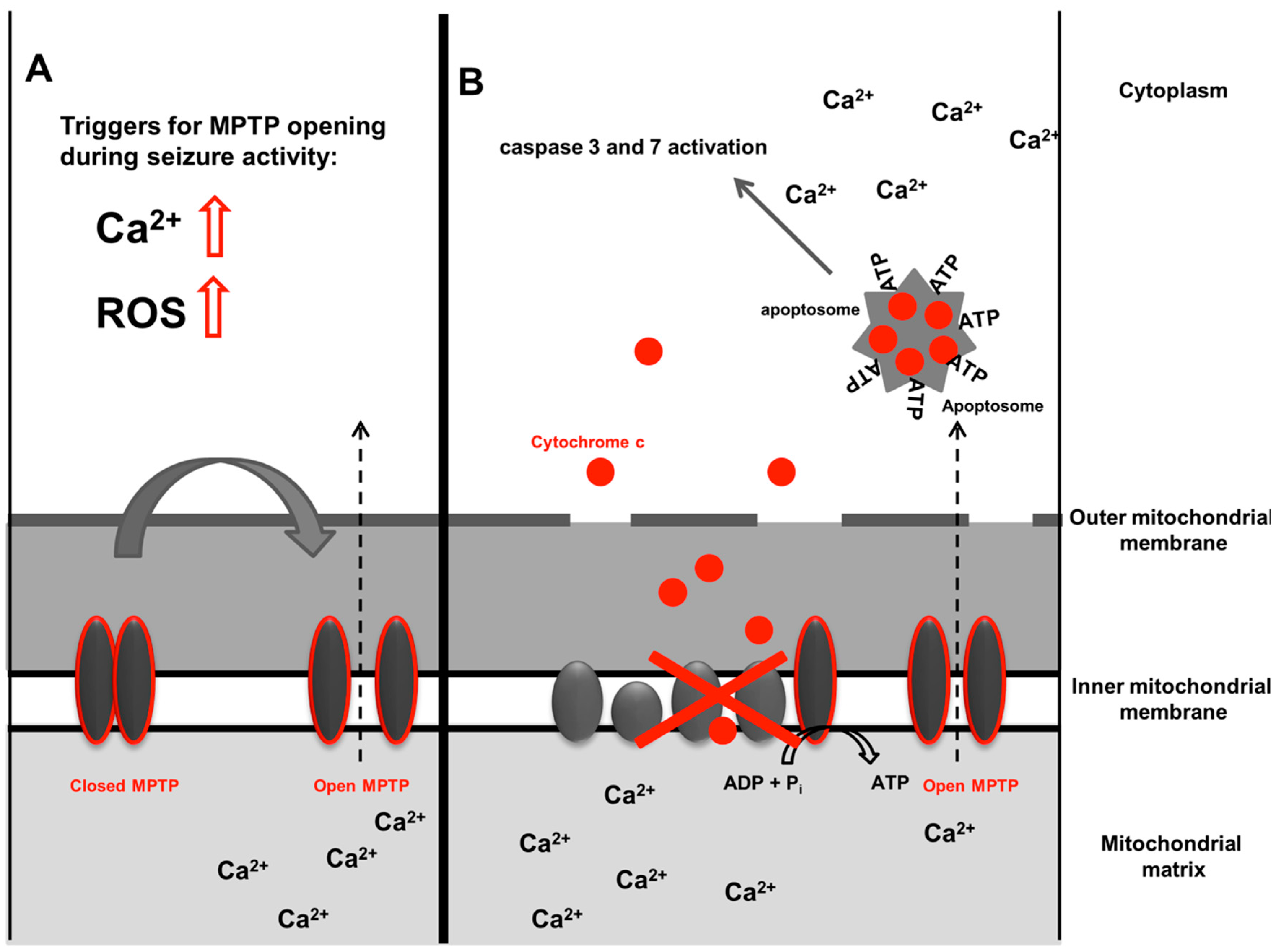
2.3. Cell Death, Mitochondria and Epilepsy
3. Excess Intracellular Ca2+ during Seizure Activity—The Endoplasmic Reticulum
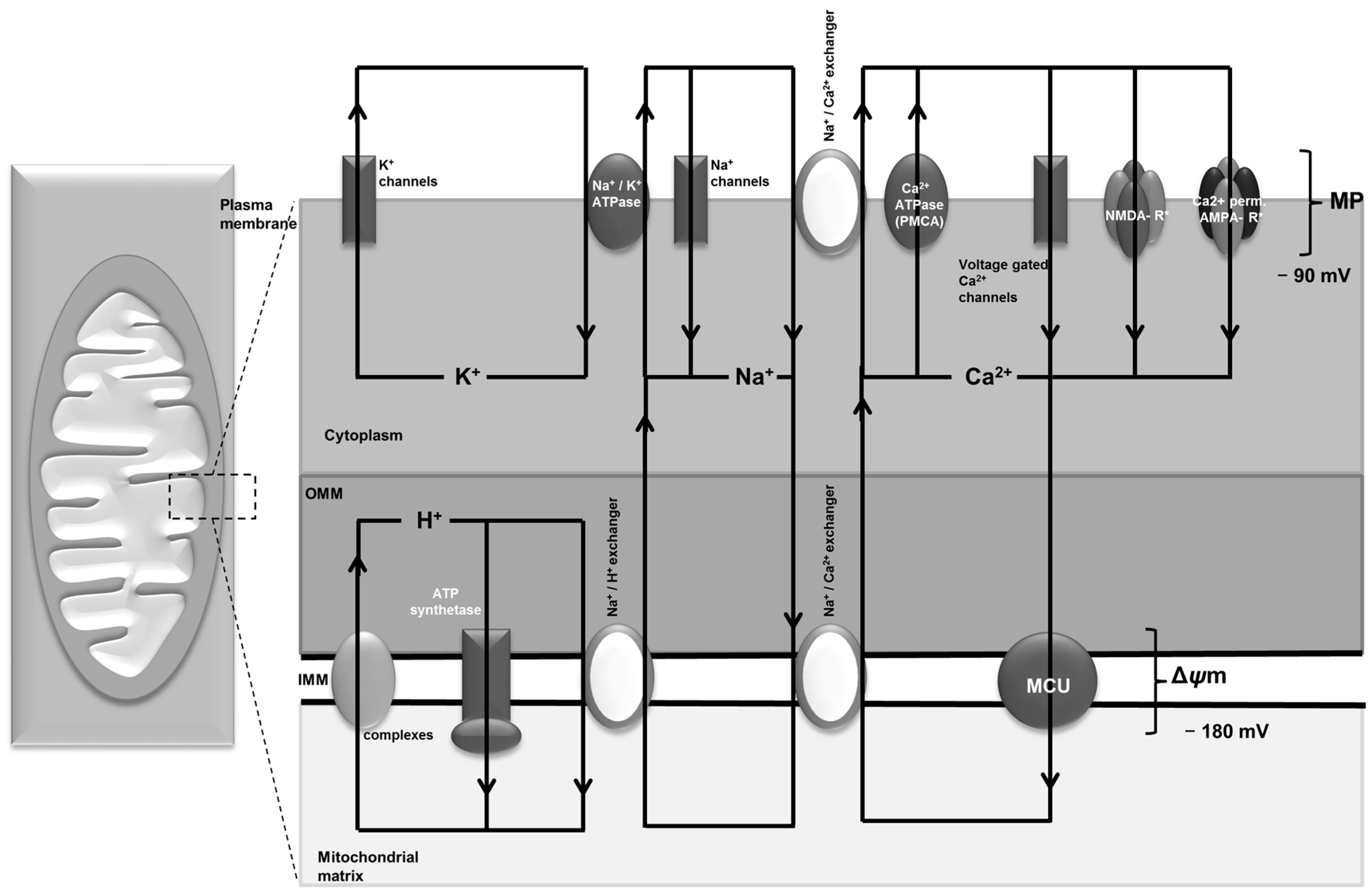
4. Ca2+ Channels and Transporters in the Plasma Cell Membrane and Their Role in Epilepsy—NMDA Receptor, AMPA Receptor, VGCC and PMCA
5. Epilepsy and Reactive Oxygen Species
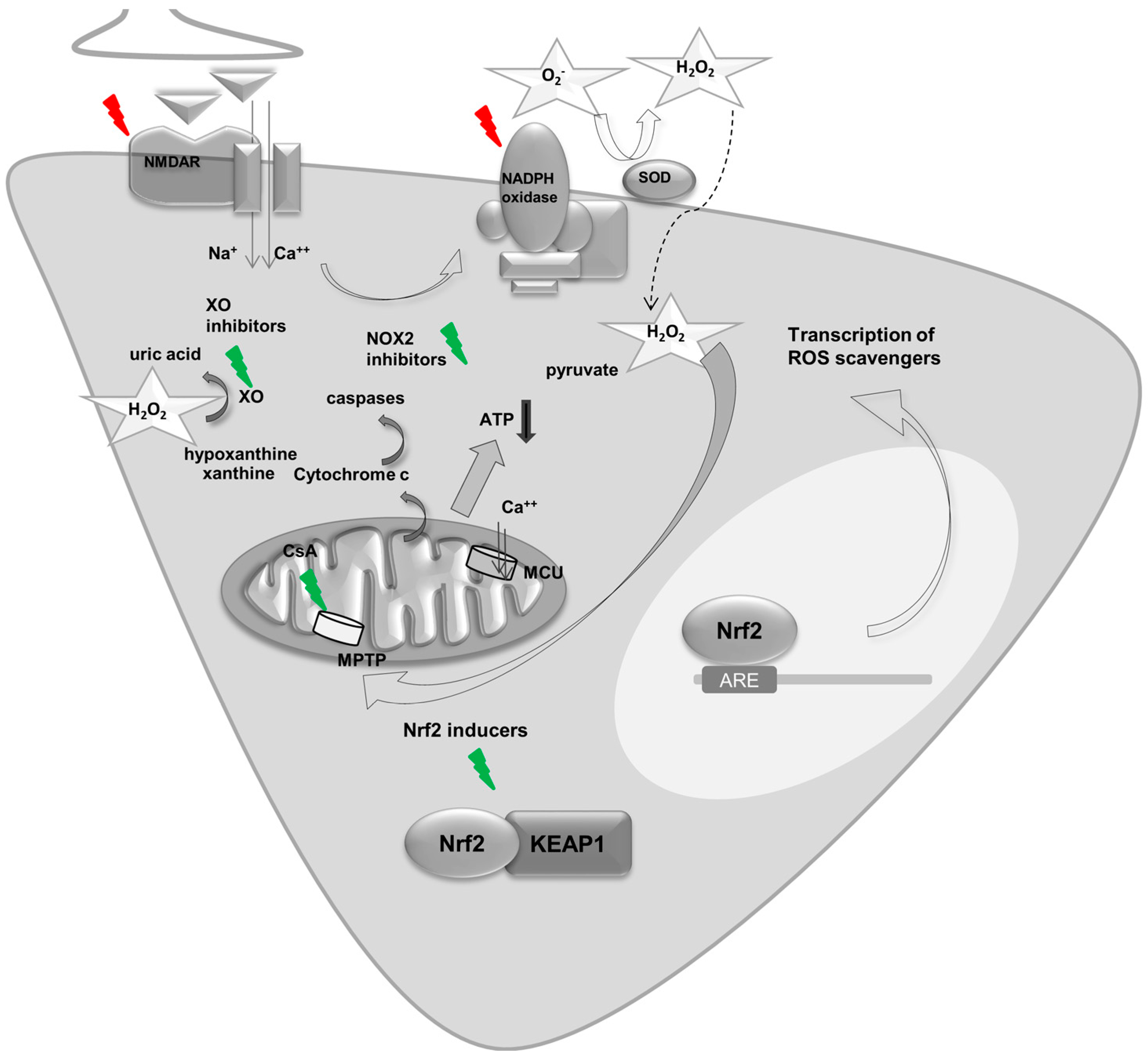
5.1. Mitochondria and ROS in Seizures and Epilepsy
5.2. NADPH Oxidase Derived ROS and Epilepsy
5.3. Other Sources of ROS in Epilepsy
6. Key Regulators of Energy Metabolism and ROS—A Focus on Nrf2 in Seizures and Epilepsy
7. Conclusions and Unmet Research Needs
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brodie, M.J.; Shorvon, S.D.; Canger, R.; Halász, P.; Johannessen, S.; Thompson, P.; Wieser, H.G.; Wolf, P. Commission on European Affairs: Appropriate standards of epilepsy care across Europe. Epilepsia 1997, 38, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Lukasiuk, K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011, 10, 173–186. [Google Scholar] [CrossRef]
- Duncan, J.S.; Sander, J.W.; Sisodiya, S.M.; Walker, M.C. Adult epilepsy. Lancet 2006, 367, 1087–1100. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Liotta, A.; Rösner, J.; Huchzermeyer, C.; Wojtowicz, A.; Kann, O.; Schmitz, D.; Heinemann, U.; Kovács, R. Energy demand of synaptic transmission at the hippocampal Schaffer-collateral synapse. J. Cereb. Blood Flow Metab. 2012, 32, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Lux, H.D.; Heinemann, U.; Dietzel, I. Ionic changes and alterations in the size of the extracellular space during epileptic activity. Adv. Neurol. 1986, 44, 619–639. [Google Scholar] [PubMed]
- Griffiths, T.; Evans, M.C.; Meldrum, B.S. Intracellular calcium accumulation in rat hippocampus during seizures induced by bicuculline or l-allylglycine. Neuroscience 1983, 10, 385–395. [Google Scholar] [CrossRef]
- Gilbert, M.E. The NMDA-receptor antagonist, MK-801, suppresses limbic kindling and kindled seizures. Brain Res. 1988, 463, 90–99. [Google Scholar] [CrossRef]
- Deshpande, L.S.; Lou, J.K.; Mian, A.; Blair, R.E.; Sombati, S.; Attkisson, E.; DeLorenzo, R.J. Time course and mechanism of hippocampal neuronal death in an in vitro model of status epilepticus: Role of NMDA receptor activation and NMDA dependent calcium entry. Eur. J. Pharmacol. 2008, 583, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Domijan, A.-M.; Walker, M.C.; Abramov, A.Y. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J. Cell Sci. 2012, 125, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Domijan, A.-M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, K.; Bai, Y.; Zhang, A.; Xia, Z.; Chao, J.; Yao, H. NADPH oxidase activation is required for pentylenetetrazole kindling-induced hippocampal autophagy. Free Radic. Biol. Med. 2016, 94, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [PubMed]
- Bernardi, P.; Petronilli, V.; Di Lisa, F.; Forte, M. A mitochondrial perspective on cell death. Trends Biochem. Sci. 2001, 26, 112–117. [Google Scholar] [CrossRef]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Sander, J.W. Some aspects of prognosis in the epilepsies: A review. Epilepsia 1993, 34, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Altmann, R. Die Elementarorganismen Und Ihre Beziehungen Zu Den Zellen; Veit & Comp.: Leipzig, Germany, 1890. [Google Scholar]
- Chance, B. Spectra and reaction kinetics of respiratory pigments of homogenized and intact cells. Nature 1952, 169, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. IV. The respiratory chain. J. Biol. Chem. 1955, 217, 429–438. [Google Scholar] [PubMed]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J. Biol. Chem. 1955, 217, 395–407. [Google Scholar] [PubMed]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Almeida, J.; Bolaños, J.P.; Moncada, S. Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proc. Natl. Acad. Sci. USA 2001, 98, 15294–15299. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology 2008, 23, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ugarte-Uribe, B.; García-Sáez, A.J. Apoptotic foci at mitochondria: In and around Bax pores. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372, 20160217. [Google Scholar] [CrossRef] [PubMed]
- King, L.J.; Lowry, O.H.; Passonneau, J.V.; Venson, V. Effects of convulsants on energy reserves in the cerebral cortex. J. Neurochem. 1967, 14, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, B.; Wilson, J.E.; Tiekert, C.G. Regulation of glycolysis in brain, in situ, during convulsions. J. Biol. Chem. 1966, 241, 5071–5075. [Google Scholar] [PubMed]
- Sanders, A.P.; Kramer, R.S.; Woodhall, B.; Currie, W.D. Brain adenosine triphosphate: Decreased concentration precedes convulsions. Science 1970, 169, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Bough, K. Energy metabolism as part of the anticonvulsant mechanism of the ketogenic diet. Epilepsia 2008, 49 (Suppl. S8), 91–93. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M. How does the ketogenic diet induce anti-seizure effects? Neurosci. Lett. 2017, 637, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Pasca, L.; De Giorgis, V.; Macasaet, J.A.; Trentani, C.; Tagliabue, A.; Veggiotti, P. The changing face of dietary therapy for epilepsy. Eur. J. Pediatr. 2016, 175, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Dressler, A.; Trimmel-Schwahofer, P.; Reithofer, E.; Mühlebner, A.; Gröppel, G.; Reiter-Fink, E.; Benninger, F.; Grassl, R.; Feucht, M. Efficacy and tolerability of the ketogenic diet in Dravet syndrome—Comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015, 109, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, E.; Ismayilova, N.; Kinali, M. Use of the Ketogenic Diet to Treat Intractable Epilepsy in Mitochondrial Disorders. J. Clin. Med. 2017, 6, E56. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, T.; Evans, M.C.; Meldrum, B.S. Intracellular sites of early calcium accumulation in the rat hippocampus during status epilepticus. Neurosci. Lett. 1982, 30, 329–334. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Chalmers, S. The integration of mitochondrial calcium transport and storage. J. Bioenerg. Biomembr. 2004, 36, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, N.; Wang, Y.; Li, Y.; Ge, X.; Wang, M. Role of the Mitochondrial Calcium Uniporter in Rat Hippocampal Neuronal Death after Pilocarpine-Induced Status Epilepticus. Neurochem. Res. 2015, 40, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S.; Vigouroux, R.A.; Brierley, J.B. Systemic factors and epileptic brain damage. Prolonged seizures in paralyzed, artificially ventilated baboons. Arch. Neurol. 1973, 29, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Schmutzhard, E.; Pfausler, B. Complications of the management of status epilepticus in the intensive care unit. Epilepsia 2011, 52 (Suppl. S8), 39–41. [Google Scholar] [CrossRef] [PubMed]
- Norwood, B.A.; Bauer, S.; Wegner, S.; Hamer, H.M.; Oertel, W.H.; Sloviter, R.S.; Rosenow, F. Electrical stimulation-induced seizures in rats: A “dose-response” study on resultant neurodegeneration. Epilepsia 2011, 52, e109–e112. [Google Scholar] [CrossRef] [PubMed]
- D’Orsi, B.; Mateyka, J.; Prehn, J.H.M. Control of mitochondrial physiology and cell death by the Bcl-2 family proteins Bax and Bok. Neurochem. Int. 2017, in press. [Google Scholar]
- Nobili, P.; Colciaghi, F.; Finardi, A.; Zambon, S.; Locatelli, D.; Battaglia, G.S. Continuous neurodegeneration and death pathway activation in neurons and glia in an experimental model of severe chronic epilepsy. Neurobiol. Dis. 2015, 83, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C.; Murphy, B.M. Modulators of neuronal cell death in epilepsy. Curr. Opin. Pharmacol. 2008, 8, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C.; Araki, T.; Schindler, C.K.; Lan, J.-Q.; Tiekoter, K.L.; Taki, W.; Simon, R.P. Activation of Bcl-2-Associated Death Protein and Counter-Response of Akt within Cell Populations during Seizure-Induced Neuronal Death. J. Neurosci. 2002, 22, 8458–8465. [Google Scholar] [PubMed]
- Henshall, D.C.; Chen, J.; Simon, R.P. Involvement of caspase-3-like protease in the mechanism of cell death following focally evoked limbic seizures. J. Neurochem. 2000, 74, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Barel, O.; Christine V Malicdan, M.; Ben-Zeev, B.; Kandel, J.; Pri-Chen, H.; Stephen, J.; Castro, I.G.; Metz, J.; Atawa, O.; Moshkovitz, S.; et al. Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain 2017, 140, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Heilbrunn, L.V. An Outline of General Physiology, 3rd ed.; W. B. Saunders: Philadelphia, PA, USA, 1952. [Google Scholar]
- Heilbrunn, L.V.; Wiercinski, F.J. The action of various cations on muscle protoplasm. J. Cell. Physiol. 1947, 29, 15–32. [Google Scholar] [CrossRef]
- Streb, H.; Irvine, R.F.; Berridge, M.J.; Schulz, I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 1983, 306, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Physiology and Pathophysiology of the Calcium Store in the Endoplasmic Reticulum of Neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef] [PubMed]
- Sokal, D.M.; Mason, R.; Parker, T.L. Multi-neuronal recordings reveal a differential effect of thapsigargin on bicuculline- or gabazine-induced epileptiform excitability in rat hippocampal neuronal networks. Neuropharmacology 2000, 39, 2408–2417. [Google Scholar] [CrossRef]
- Rutecki, P.A.; Sayin, U.; Yang, Y.; Hadar, E. Determinants of ictal epileptiform patterns in the hippocampal slice. Epilepsia 2002, 43 (Suppl. S5), 179–183. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Kanemaru, K.; Okubo, Y.; Nakaune, T.; Suzuki, J.; Shibata, K.; Sugiyama, H.; Koyama, R.; Murayama, T.; Ito, A.; et al. Nitric Oxide-induced Activation of the Type 1 Ryanodine Receptor Is Critical for Epileptic Seizure-induced Neuronal Cell Death. EBioMedicine 2016, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Ormandy, G.C.; Jope, R.S.; Snead, O.C. Anticonvulsant actions of MK-801 on the lithium-pilocarpine model of status epilepticus in rats. Exp. Neurol. 1989, 106, 172–180. [Google Scholar] [CrossRef]
- Fujikawa, D.G.; Daniels, A.H.; Kim, J.S. The competitive NMDA receptor antagonist CGP 40116 protects against status epilepticus-induced neuronal damage. Epilepsy Res. 1994, 17, 207–219. [Google Scholar] [CrossRef]
- Finardi, A.; Colciaghi, F.; Castana, L.; Locatelli, D.; Marras, C.E.; Nobili, P.; Fratelli, M.; Bramerio, M.A.; Lorusso, G.; Battaglia, G.S. Long-duration epilepsy affects cell morphology and glutamatergic synapses in type IIB focal cortical dysplasia. Acta Neuropathol. 2013, 126, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Colciaghi, F.; Finardi, A.; Nobili, P. Intrinsic epileptogenicity of dysplastic cortex: Converging data from experimental models and human patients. Epilepsia 2013, 54 (Suppl. S6), 33–36. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Tüzün, E.; Wu, H.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-d-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blümcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135, 1622–1638. [Google Scholar] [CrossRef] [PubMed]
- Camdessanché, J.-P.; Streichenberger, N.; Cavillon, G.; Rogemond, V.; Jousserand, G.; Honnorat, J.; Convers, P.; Antoine, J.-C. Brain immunohistopathological study in a patient with anti-NMDAR encephalitis. Eur. J. Neurol. 2011, 18, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Tüzün, E.; Zhou, L.; Baehring, J.M.; Bannykh, S.; Rosenfeld, M.R.; Dalmau, J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 2009, 118, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium; Epilepsy Phenome/Genome Project; Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Ohba, C.; Shiina, M.; Tohyama, J.; Haginoya, K.; Lerman-Sagie, T.; Okamoto, N.; Blumkin, L.; Lev, D.; Mukaida, S.; Nozaki, F.; et al. GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia 2015, 56, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.E.; Inada, K.; Koller, B.H.; Moy, S.S. Increased sensitivity to kainic acid in a genetic model of reduced NMDA receptor function. Brain Res. 2010, 1307, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Gorji, A.; Speckmann, E.J. Low concentration of dl-2-amino-5-phosphonovalerate induces epileptiform activity in guinea pig hippocampal slices. Epilepsia 2001, 42, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, K.; Todorovic, M.; Kapur, J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann. Neurol. 2012, 72, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zamponi, G.W. Voltage-Gated Calcium Channels and Idiopathic Generalized Epilepsies. Physiol. Rev. 2006, 86, 941–966. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.M.; Snutch, T.P. Voltage-Gated Calcium Channels in Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Van Loo, K.M.J.; Schaub, C.; Pitsch, J.; Kulbida, R.; Opitz, T.; Ekstein, D.; Dalal, A.; Urbach, H.; Beck, H.; Yaari, Y.; et al. Zinc regulates a key transcriptional pathway for epileptogenesis via metal-regulatory transcription factor 1. Nat. Commun. 2015, 6, 8688. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.J.; Pitsch, J.; Sochivko, D.; Opitz, T.; Staniek, M.; Chen, C.-C.; Campbell, K.P.; Schoch, S.; Yaari, Y.; Beck, H. Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J. Neurosci. 2008, 28, 13341–13353. [Google Scholar] [CrossRef] [PubMed]
- Naziroğlu, M.; Kutluhan, S.; Yilmaz, M. Selenium and topiramate modulates brain microsomal oxidative stress values, Ca2+-ATPase activity, and EEG records in pentylentetrazol-induced seizures in rats. J. Membr. Biol. 2008, 225, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ketelaars, S.O.M.; Gorter, J.A.; Aronica, E.; Wadman, W.J. Calcium extrusion protein expression in the hippocampal formation of chronic epileptic rats after kainate-induced status epilepticus. Epilepsia 2004, 45, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Murray, K.D.; Garcia, V.B.; Strehler, E.E.; Isackson, P.J. Seizure-induced alterations of plasma membrane calcium ATPase isoforms 1, 2 and 3 mRNA and protein in rat hippocampus. Brain Res. Mol. Brain Res. 1997, 45, 230–238. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Malinska, D.; Kulawiak, B.; Kudin, A.P.; Kovacs, R.; Huchzermeyer, C.; Kann, O.; Szewczyk, A.; Kunz, W.S. Complex III-dependent superoxide production of brain mitochondria contributes to seizure-related ROS formation. Biochim. Biophys. Acta 2010, 1797, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Pestana, R.R.F.; Kinjo, E.R.; Hernandes, M.S.; Britto, L.R.G. Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 2010, 484, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.T.; Hergenrother, P.J. Poly(ADP-ribose) makes a date with death. Curr. Opin. Chem. Biol. 2007, 11, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-C.; Baker, J.S.; Donti, T.; Graham, B.H.; Craigen, W.J.; Anderson, A.E. Mitochondrial Dysfunction Mediated by Poly(ADP-Ribose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus. Int. J. Mol. Sci. 2017, 18, E1502. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C.; Halliwell, B. The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem. Sci. 1990, 15, 129–135. [Google Scholar] [CrossRef]
- Kovac, S.; Dinkova-Kostova, A.T.; Abramov, A.Y. The Role of Reactive Oxygen Species in Epilepsy. React. Oxyg. Species 2016, 1, 38–52. [Google Scholar] [CrossRef]
- Bruce, A.J.; Baudry, M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic. Biol. Med. 1995, 18, 993–1002. [Google Scholar] [CrossRef]
- Kovács, R.; Schuchmann, S.; Gabriel, S.; Kann, O.; Kardos, J.; Heinemann, U. Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J. Neurophysiol. 2002, 88, 2909–2918. [Google Scholar] [CrossRef] [PubMed]
- Delanty, N.; Dichter, D.M. Antioxidant therapy in neurologic disease. Arch. Neurol. 2000, 57, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Volmering, E.; Niehusmann, P.; Peeva, V.; Grote, A.; Zsurka, G.; Altmüller, J.; Nürnberg, P.; Becker, A.J.; Schoch, S.; Elger, C.E.; et al. Neuropathological signs of inflammation correlate with mitochondrial DNA deletions in mesial temporal lobe epilepsy. Acta Neuropathol. 2016, 132, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Royer-Pokora, B.; Kunkel, L.M.; Monaco, A.P.; Goff, S.C.; Newburger, P.E.; Baehner, R.L.; Cole, F.S.; Curnutte, J.T.; Orkin, S.H. Cloning the gene for an inherited human disorder—Chronic granulomatous disease—On the basis of its chromosomal location. Nature 1986, 322, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Jaquet, V.; Krause, K.-H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [PubMed]
- Olguín-Albuerne, M.; Domínguez, G.; Morán, J. Effect of staurosporine in the morphology and viability of cerebellar astrocytes: Role of reactive oxygen species and NADPH oxidase. Oxidative Med. Cell. Longev. 2014, 2014, 678371. [Google Scholar] [CrossRef] [PubMed]
- Hohn, D.C.; Lehrer, R.I. NADPH oxidase deficiency in X-linked chronic granulomatous disease. J. Clin. Investig. 1975, 55, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Nishimura, A.; Nakamura, K.; Matsuo, R.; Wakisaka, Y.; Kamouchi, M.; Kitazono, T. Nox4 is a major source of superoxide production in human brain pericytes. J. Vasc. Res. 2014, 51, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Sirokmány, G.; Donkó, Á.; Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol. Sci. 2016, 37, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Girouard, H.; Wang, G.; Gallo, E.F.; Anrather, J.; Zhou, P.; Pickel, V.M.; Iadecola, C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 2009, 29, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Li, Q.-Y.; Chang, L.-Y.; Crapo, J.; Liang, L.-P. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J. Neurochem. 2005, 92, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Mastroberardino, P.G.; Hu, X.; Montero, L.; Greenamyre, J.T. Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol. Dis. 2011, 42, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Chan, J.Y.H.; Hsu, K.; Chang, A.Y.W.; Chan, S.H.H. Brain-Derived Neurotrophic Factor Ameliorates Brain Stem Cardiovascular Dysregulation during Experimental Temporal Lobe Status Epilepticus. PLoS ONE 2012, 7, e33527. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Ryu, H.J.; Kang, T.-C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/ endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Natrella, F.; Belmonte, G.; Miracco, C.; Cervellati, F.; Ciccoli, L.; Mariottini, A.; Rocchi, R.; Vatti, G.; Bua, A.; et al. NADPH oxidase activation and 4-hydroxy-2-nonenal/aquaporin-4 adducts as possible new players in oxidative neuronal damage presents in drug-resistant epilepsy. Biochim. Biophys. Acta 2015, 1852, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Hamil, N.; Abramov, A.Y.; Walker, M.C.; Kovac, S. Status epilepticus results in persistent overproduction of reactive oxygen species, inhibition of which is neuroprotective. Neuroscience 2015, 303, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J.; Li, S.; Basu, U.; Tian, J.; Zimmerman, M.C. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H19–H28. [Google Scholar] [CrossRef] [PubMed]
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Dürr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Yokoyama, H.; Niwa, R.; Konaka, R.; Ohya-Nishiguchi, H.; Kamada, H. Generation of lipid radicals in the hippocampal extracellular space during kainic acid-induced seizures in rats. Epilepsy Res. 1997, 26, 329–333. [Google Scholar] [CrossRef]
- Baran, H.; Vass, K.; Lassmann, H.; Hornykiewicz, O. The cyclooxygenase and lipoxygenase inhibitor BW755C protects rats against kainic acid-induced seizures and neurotoxicity. Brain Res. 1994, 646, 201–206. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Baird, L.; Holmström, K.M.; Meyer, C.J.; Abramov, A.Y. The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.; Zeng, W.; Hus, J.-C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Kita, M.; Cohan, S.L.; Henson, L.J.; Zambrano, J.; Scannevin, R.H.; O’Gorman, J.; Novas, M.; Dawson, K.T.; Phillips, J.T. BG-12 (dimethyl fumarate): A review of mechanism of action, efficacy, and safety. Curr. Med. Res. Opin. 2014, 30, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of γ-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Shafer, J.; Fiskum, G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic. Biol. Med. 2011, 51, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Aparicio, L.; Pérez-Cruz, C.; Zavala-Tecuapetla, C.; Granados-Rojas, L.; Rivera-Espinosa, L.; Montesinos-Correa, H.; Hernández-Damián, J.; Pedraza-Chaverri, J.; Sampieri, A.; Coballase-Urrutia, E.; et al. Overview of Nrf2 as Therapeutic Target in Epilepsy. Int. J. Mol. Sci. 2015, 16, 18348–18367. [Google Scholar] [CrossRef] [PubMed]
- Mazzuferi, M.; Kumar, G.; van Eyll, J.; Danis, B.; Foerch, P.; Kaminski, R.M. Nrf2 defense pathway: Experimental evidence for its protective role in epilepsy. Ann. Neurol. 2013, 74, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pozo, C.; Tan, K.N.; Borges, K. Sulforaphane is anticonvulsant and improves mitochondrial function. J. Neurochem. 2015, 135, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A.; Pascente, R.; Liang, L.-P.; Villa, B.R.; et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2017, 140, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Nieoczym, D.; Kowalczuk-Vasilev, E.; Wyska, E.; Wlaź, P. Increased seizure susceptibility and other toxicity symptoms following acute sulforaphane treatment in mice. Toxicol. Appl. Pharmacol. 2017, 326, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Williamson, T.P.; Amirahmadi, S.; Joshi, G.; Kaludov, N.K.; Martinov, M.N.; Johnson, D.A.; Johnson, J.A. Discovery of potent, novel Nrf2 inducers via quantum modeling, virtual screening and in vitro experimental validation. Chem. Biol. Drug Des. 2012, 80, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study | Species | Epilepsy Model (In Vivo/Ex Vivo/In Vitro) | NADPH Oxidase Subtype Studied | NADPH Oxidase Inhibition | Main Findings |
|---|---|---|---|---|---|
| Zhu et al., 2016 [14] | Mouse | Pentylenetetrazol (PTZ) model (in vivo) | No | Pharmacological (Apocynin) |
|
| Williams et al., 2015, [118] | Rat | Perforant path stimulation (PPS) model (ex vivo and in vivo) | No | Pharmacological (AEBSF) |
|
| Pecorelli et al., 2015, [117] | Human | Tissue from Patients with drug resistant epilepsy (ex vivo) | Yes (NOX2) | N/A |
|
| Kovac et al., 2015 [13] | Rat | Low magnesium model (in vitro) | Yes (NOX2) | Pharmacological (AEBSF, gp-91-tat) |
|
| Kim et al., 2013, [116] | Rat | Pilocarpine induced SE (in vivo) | Yes (NOX2) | Pharmacological (Apocynin) |
|
| Kim et al., 2013, [112] | Rat | Pilocarpine induced epilepsy (ex vivo and in vivo) | Yes (NOX2) | Pharmacological (Apocynin) |
|
| Tsai et al., 2012, [115] | Rat | SE due to focal temporal injection of kainic acid (TLSE; ex vivo and in vivo) | Yes (NOX2) | Pharmacological (Apocynin) |
|
| Di Maio et al., 2011, [114] | Rat | Pilocarpine induced seizures (in vitro and ex vivo) | Yes (NOX2) | Pharmacological (Apocynin, 6-amino-nicotidamid) |
|
| Pestana et al., 2010, [91] | Rat | Pilocarpine induced SE (in vivo) | No | Pharmacological (Apocynin) |
|
| Patel et al., 2005, [113] | Rat | Kainate model of epilepsy (ex vivo) | Yes (NOX2) | N/A |
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovac, S.; Dinkova Kostova, A.T.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species. Int. J. Mol. Sci. 2017, 18, 1935. https://doi.org/10.3390/ijms18091935
Kovac S, Dinkova Kostova AT, Herrmann AM, Melzer N, Meuth SG, Gorji A. Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species. International Journal of Molecular Sciences. 2017; 18(9):1935. https://doi.org/10.3390/ijms18091935
Chicago/Turabian StyleKovac, Stjepana, Albena T. Dinkova Kostova, Alexander M. Herrmann, Nico Melzer, Sven G. Meuth, and Ali Gorji. 2017. "Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species" International Journal of Molecular Sciences 18, no. 9: 1935. https://doi.org/10.3390/ijms18091935
APA StyleKovac, S., Dinkova Kostova, A. T., Herrmann, A. M., Melzer, N., Meuth, S. G., & Gorji, A. (2017). Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species. International Journal of Molecular Sciences, 18(9), 1935. https://doi.org/10.3390/ijms18091935