Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
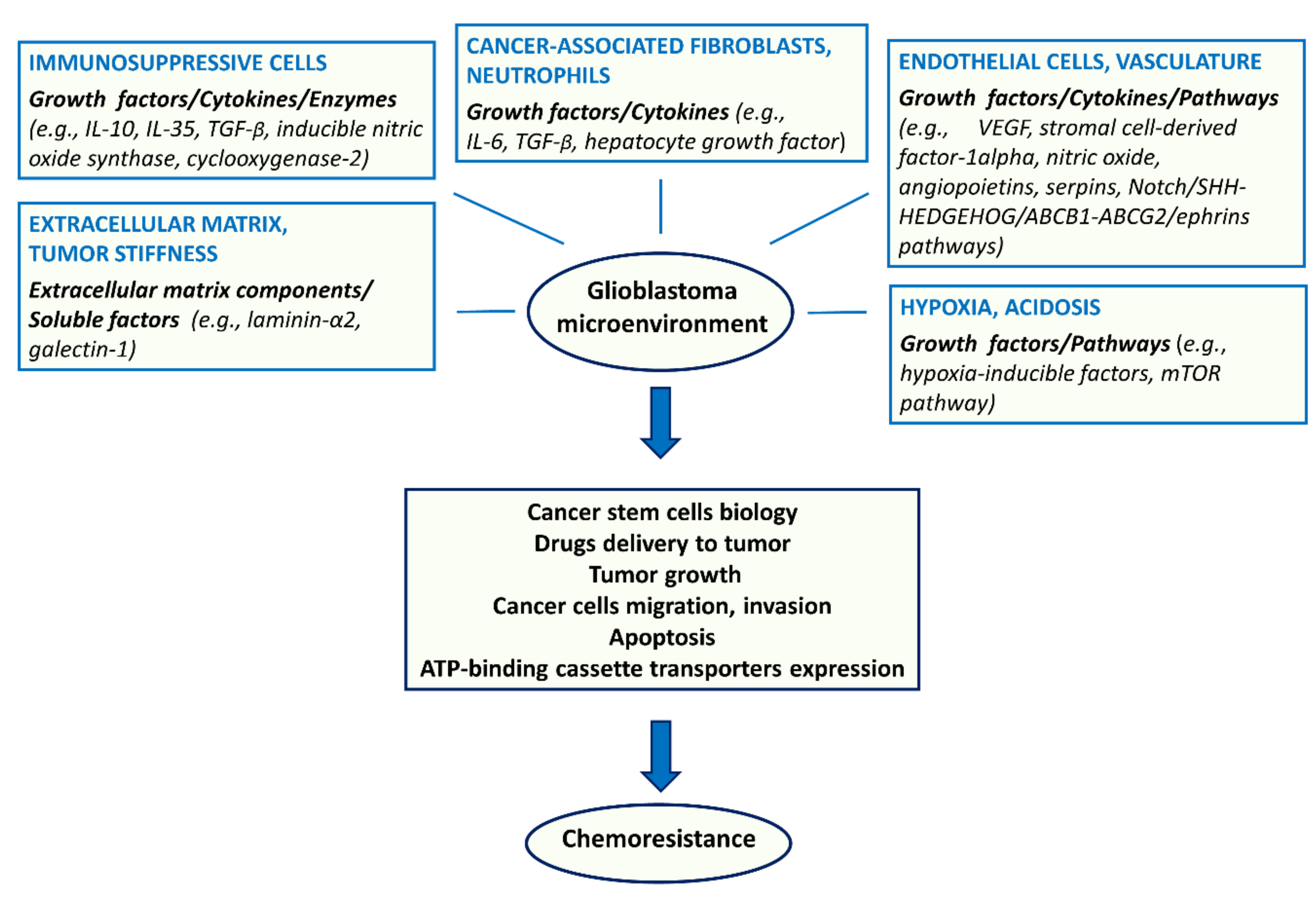
2. Chemoresistance Due to Changes in TME
2.1. Endothelial Cells
2.2. Tumor-Associated Immunosuppressive Cells
2.3. ECM
2.4. Hypoxia
2.5. Acidosis
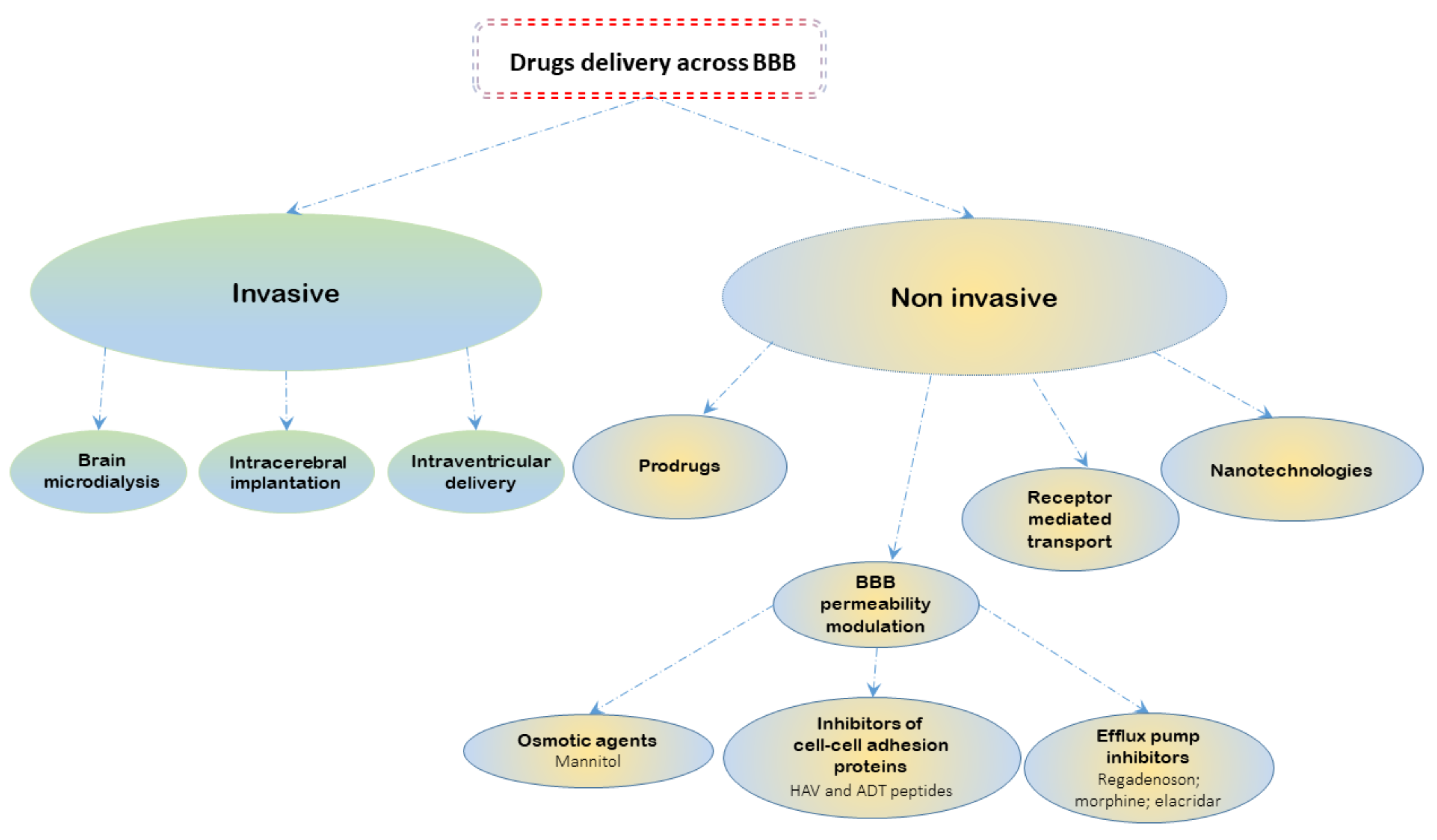
3. BBB Pharmacological Modulation for Treatment of Human Glioblastoma
3.1. Paracellular Modulation
3.2. Transcellular Modulation
4. Genetic Aspects of Chemoresistance in Glioblastoma
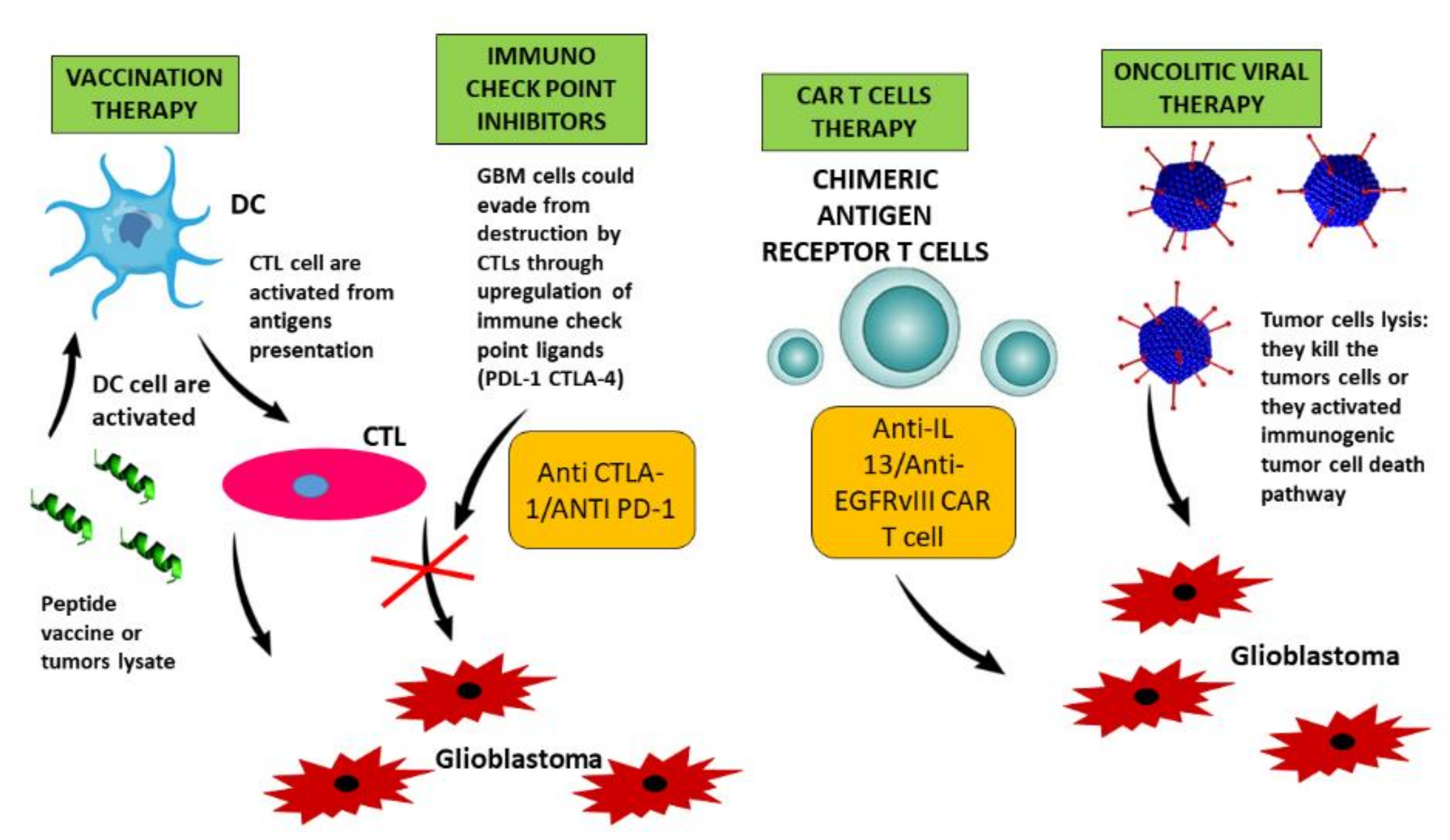
5. Immunotherapy in Glioblastoma: Overcoming Barriers to Response
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| ABCG2 | ATP binding cassette subfamily G member 2 |
| ADT | Ala-Asp-Thr |
| BBB | blood brain barrier |
| BCRP | breast cancer resistance protein |
| BER | base excision repair |
| CAFs | cancer-associated fibroblasts |
| CAR | chimeric antigen receptor |
| CNS | central nervous system |
| CSCs | cancer stem cells |
| CTLs | cytotoxic T lymphocytes |
| DCs | dendritic cells |
| DDR | DNA damage |
| Dox | doxorubicin |
| DSBs | double-strand breaks |
| ECM | extracellular matrix |
| EGFRvIII | EGFR variant III |
| GdDTPA | gadolinium-DTPA |
| GSCs | glioblastoma stem cells |
| HAV | His-Ala-Val |
| HIFs | hypoxia-inducible factors |
| HR | homologous recombination |
| IDO | indolamine 2,3 dioxygenase |
| IL-13Rα2 | IL-13 receptor subunit-α2 |
| IR | ionizing radiation |
| MDCK | madin-darby canine kidney |
| MDR | multi drug resistance |
| MDR1 | multi drug resistance 1 |
| MDSCs | myeloid-derived suppressor cells |
| MGMT | O6-methylguanine DNA methyltransferase |
| MHC | major histocompatibility complex |
| miRNAs | microRNAs |
| MMR | mismatch repair system |
| MRP1 | multi drug resistance-associated protein 1 |
| mTOR | mammalian target of rapamycin |
| NDRG1 | N-myc downstream regulated gene 1 |
| NHEJ | non-homologous end joining |
| NK | natural killer |
| PD-1 | programmed cell death protein-1 |
| PEG | polyethylene glycol |
| P-gp | P-glycoprotein |
| R800 | rhodamine 800 |
| ROS | reactive oxygen species |
| SSBs | single-strand breaks |
| TAMs | tumor-associated macrophages |
| TEER | transepithelial electrical resistance |
| TME | tumor microenvironment |
| TMZ | temozolomide |
| Treg | T-regulatory |
| VEGF | vascular endothelial growth factor |
References
- Sonnenschein, C.; Soto, A.M. Theories of carcinogenesis: An emerging perspective. Semin. Cancer Biol. 2008, 18, 372–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Swanton, C. How darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 2011, 11, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Qin, Z.; Chen, Z.; Xie, L.; Wang, R.; Zhao, H. Tumor microenvironment in treatment of glioma. Open Med. (Wars) 2017, 12, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, R.L. Brain tumor hypoxia: Tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J. Neurooncol. 2009, 92, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Aspects Med. 2017, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Persano, L.; Rampazzo, E.; Basso, G.; Viola, G. Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem. Pharmacol. 2013, 85, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shervington, A. Chemoresistance in gliomas. Mol. Cell. Biochem. 2008, 312, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Bredel, M. Anticancer drug resistance in primary human brain tumors. Brain Res. Rev. 2001, 35, 161–204. [Google Scholar] [CrossRef]
- Papademetriou, I.T.; Porter, T. Promising approaches to circumvent the blood-brain barrier: Progress, pitfalls and clinical prospects in brain cancer. Ther. Deliv. 2015, 6, 989–1016. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Ruzevick, J.; Phallen, J.; Belcaid, Z.; Lim, M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin. Dev. Immunol. 2011, 2011, 732413. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Seano, G. Targeting the perivascular niche in brain tumors. Curr. Opin. Oncol. 2018, 30, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer stem cells and their microenvironment: Biology and therapeutic implications. Stem. Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma To target the tumor cell or the microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 16; pp. 315–340. ISBN 978-0-9944381-2-6. [Google Scholar]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma stem cells: Signaling, microenvironment, and therapy. Stem. Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Del Bufalo, D.; Cinà, I.; Di Benedetto, A.; Mottolese, M.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. The impact of the tumor microenvironment on the properties of glioma stem-like cells. Front. Oncol. 2017, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory t cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory t cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of foxp3+ regulatory t cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Hiraoka, N.; Yamagami, W.; Ojima, H.; Kanai, Y.; Kosuge, T.; Nakajima, A.; Hirohashi, S. Foxp3+ regulatory t cells affect the development and progression of hepatocarcinogenesis. Clin. Cancer Res. 2007, 13, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Kawaida, H.; Takahashi, A.; Sugai, H.; Mimura, K.; Miyagawa, N.; Omata, H.; Fujii, H. Cd4(+)cd25high regulatory t cells increase with tumor stage in patients with gastric and esophageal cancers. Cancer Immunol. Immunother. 2006, 55, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Trylcova, J.; Busek, P.; Smetana, K.; Balaziova, E.; Dvorankova, B.; Mifkova, A.; Sedo, A. Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumour. Biol. 2015, 36, 5873–5879. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, I.J.; Iravani, M.; Popov, S.; Robertson, D.; Al-Sarraj, S.; Jones, C.; Isacke, C.M. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS ONE 2010, 5, e9808. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.R.; Huang, P.; et al. Laminin α 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Corsini, N.S.; Martin-Villalba, A. Integrin α 6: Anchors away for glioma stem cells. Cell Stem Cell 2010, 6, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1α-tenascin c feedback to regulate glioblastoma aggression. Nat. Cell. Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Tse, A.P.; Huang, Y.P.; Zhu, Y.T.; Chiu, D.K.; Lai, R.K.; Au, S.L.; Kai, A.K.; Lee, J.M.; Wei, L.L.; et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014, 60, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, M.F.; Siedlik, M.J.; Han, S.; Stallings-Mann, M.; Radisky, D.C.; Nelson, C.M. Tissue stiffness and hypoxia modulate the integrin-linked kinase ilk to control breast cancer stem-like cells. Cancer Res. 2016, 76, 5277–5287. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Mishra, D.P. Matrix reloaded: Ccn, tenascin and sibling group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhi, W.I.; Lu, H.; Samanta, D.; Chen, I.; Gabrielson, E.; Semenza, G.L. Hypoxia-inducible factors regulate pluripotency factor expression by znf217- and alkbh5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 2016, 7, 64527–64542. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.M.; Dai, Y.; Vieweg, J.; Siemann, D.W. Hypoxia regulates sox2 expression to promote prostate cancer cell invasion and sphere formation. Am. J. Cancer Res. 2016, 6, 1078–1088. [Google Scholar] [PubMed]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-independent, cd133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell. Oncol. (Dordr.) 2018, 41, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Baraldi, P.G.; Borea, P.A. Hypoxia inhibits paclitaxel-induced apoptosis through adenosine-mediated phosphorylation of bad in glioblastoma cells. Mol. Pharmacol. 2007, 72, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Lin, Y.J.; Wu, C.P.; Lee, H.T.; Shyu, W.C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Pantisano, V.; Puca, R.; Porru, M.; Aiello, A.; Grasselli, A.; Leonetti, C.; Safran, M.; Rechavi, G.; Givol, D.; et al. Zinc downregulates hif-1α and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE 2010, 5, e15048. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feng, P.; Li, S.; Long, D.; Cheng, J.; Lu, Y.; Zhou, D. Effect of hypoxia-inducible factor-1α silencing on the sensitivity of human brain glioma cells to doxorubicin and etoposide. Neurochem. Res. 2009, 34, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.; Catacuzzeno, L.; Sforna, L.; Mangino, G.; Carlomagno, S.; Mincione, G.; Petrozza, V.; Ragona, G.; Franciolini, F.; Calogero, A. Bk channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell. Physiol. 2018, 233, 6866–6877. [Google Scholar] [CrossRef] [PubMed]
- Weiler, M.; Blaes, J.; Pusch, S.; Sahm, F.; Czabanka, M.; Luger, S.; Bunse, L.; Solecki, G.; Eichwald, V.; Jugold, M.; et al. Mtor target ndrg1 confers mgmt-dependent resistance to alkylating chemotherapy. Proc. Natl. Acad. Sci. USA 2014, 111, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppicelli, S.; Andreucci, E.; Ruzzolini, J.; Laurenzana, A.; Margheri, F.; Fibbi, G.; Del Rosso, M.; Bianchini, F.; Calorini, L. The acidic microenvironment as a possible niche of dormant tumor cells. Cell. Mol. Life Sci. 2017, 74, 2761–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: Adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front. Immunol. 2017, 8, 1887. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, A.S.; Alexander, M.; Kahlon, A.; Wright, J. Lactate levels with glioblastoma multiforme. Proc. (Bayl. Univ. Med. Cent.) 2016, 29, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Brizel, D.M.; Schroeder, T.; Scher, R.L.; Walenta, S.; Clough, R.W.; Dewhirst, M.W.; Mueller-Klieser, W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 349–353. [Google Scholar] [CrossRef]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell. Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by γ-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppicelli, S.; Bianchini, F.; Torre, E.; Calorini, L. Contribution of acidic melanoma cells undergoing epithelial-to-mesenchymal transition to aggressiveness of non-acidic melanoma cells. Clin. Exp. Metast. 2014, 31, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Raghunand, N.; Gillies, R.J. Ph and drug resistance in tumors. Drug Resist. Updat. 2000, 3, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for cns drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Pafundi, D.H.; Laack, N.N.; Youland, R.S.; Parney, I.F.; Lowe, V.J.; Giannini, C.; Kemp, B.J.; Grams, M.P.; Morris, J.M.; Hoover, J.M.; et al. Biopsy validation of 18f-dopa pet and biodistribution in gliomas for neurosurgical planning and radiotherapy target delineation: Results of a prospective pilot study. Neuro-Oncology 2013, 15, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Onda, K.; Tanaka, R.; Takahashi, H.; Takeda, N.; Ikuta, F. Cerebral glioblastoma with cerebrospinal fluid dissemination: A clinicopathological study of 14 cases examined by complete autopsy. Neurosurgery 1989, 25, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A. Blood-brain barrier permeability considerations for cns-targeted compound library design. Curr. Opin. Chem. Biol. 2008, 12, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, R.J.; Carpenter, K.L.; Guilfoyle, M.R.; Helmy, A.; Hutchinson, P.J. Cerebral microdialysis in clinical studies of drugs: Pharmacokinetic applications. J. Pharmacokinet. Pharmacodyn. 2013, 40, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Yang, T.C.; Yang, S.T.; Chen, Y.C.; Tseng, Y.Y. Biodegradable hybrid-structured nanofibrous membrane supported chemoprotective gene therapy enhances chemotherapy tolerance and efficacy in malignant glioma rats. Artif. Cells Nanomed. Biotechnol. 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pompe, R.S.; von Bueren, A.O.; Mynarek, M.; von Hoff, K.; Friedrich, C.; Kwiecien, R.; Treulieb, W.; Lindow, C.; Deinlein, F.; Fleischhack, G.; et al. Intraventricular methotrexate as part of primary therapy for children with infant and/or metastatic medulloblastoma: Feasibility, acute toxicity and evidence for efficacy. Eur. J. Cancer 2015, 51, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.A.; Raghavan, S.; Baskin, D.S. Pam-obg: A monoamine oxidase b specific prodrug that inhibits mgmt and generates dna interstrand crosslinks, potentiating temozolomide and chemoradiation therapy in intracranial glioblastoma. Oncotarget 2018, 9, 23923–23943. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.A.; Lopez Espinoza, F.; Mendoza, D.; Kato, Y.; Inagaki, A.; Hiraoka, K.; Kasahara, N.; Gruber, H.E.; Jolly, D.J.; Robbins, J.M. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro. Oncol. 2017, 19, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.; Colombeau, L.; Gries, M.; Peterlini, T.; Mathieu, C.; Thomas, N.; Boura, C.; Frochot, C.; Vanderesse, R.; Lux, F.; et al. Ultrasmall aguix theranostic nanoparticles for vascular-targeted interstitial photodynamic therapy of glioblastoma. Int. J. Nanomed. 2017, 12, 7075–7088. [Google Scholar] [CrossRef] [PubMed]
- Nafee, N.; Gouda, N. Nucleic acids-based nanotherapeutics crossing the blood brain barrier. Curr. Gene Ther. 2017, 17, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim. Biophys. Act. 2009, 1788, 892–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charest, G.; Sanche, L.; Fortin, D.; Mathieu, D.; Paquette, B. Optimization of the route of platinum drugs administration to optimize the concomitant treatment with radiotherapy for glioblastoma implanted in the fischer rat brain. J. Neurooncol. 2013, 115, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, D.J.; Doolittle, N.D.; Gahramanov, S.; Hedrick, N.A.; Delashaw, J.B.; Neuwelt, E.A. Intra-arterial chemotherapy with osmotic blood-brain barrier disruption for aggressive oligodendroglial tumors: Results of a phase i study. Neurosurgery 2010, 66, 48–58; discussion 58. [Google Scholar] [CrossRef] [PubMed]
- Hiesiger, E.M.; Voorhies, R.M.; Basler, G.A.; Lipschutz, L.E.; Posner, J.B.; Shapiro, W.R. Opening the blood-brain and blood-tumor barriers in experimental rat brain tumors: The effect of intracarotid hyperosmolar mannitol on capillary permeability and blood flow. Ann. Neurol. 1986, 19, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Fukui, M.; Nishio, S.; Kitamura, K.; Nagara, H. Hyperosmotic blood-brain barrier disruption in brains of rats with an intracerebrally transplanted rg-c6 tumor. J. Neurosurg. 1987, 66, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Boockvar, J.A.; Tsiouris, A.J.; Hofstetter, C.P.; Kovanlikaya, I.; Fralin, S.; Kesavabhotla, K.; Seedial, S.M.; Pannullo, S.C.; Schwartz, T.H.; Stieg, P.; et al. Safety and maximum tolerated dose of superselective intraarterial cerebral infusion of bevacizumab after osmotic blood-brain barrier disruption for recurrent malignant glioma. Clinical article. J. Neurosurg. 2011, 114, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Emerich, D.F. Facilitation of drug entry into the cns via transient permeation of blood brain barrier: Laboratory and preliminary clinical evidence from bradykinin receptor agonist, cereport. Brain Res. Bull. 2003, 60, 297–306. [Google Scholar] [CrossRef]
- Inamura, T.; Black, K.L. Bradykinin selectively opens blood-tumor barrier in experimental brain tumors. J. Cereb. Blood Flow Metab. 1994, 14, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Makagiansar, I.T.; Avery, M.; Hu, Y.; Audus, K.L.; Siahaan, T.J. Improving the selectivity of hav-peptides in modulating e-cadherin-e-cadherin interactions in the intercellular junction of mdck cell monolayers. Pharm. Res. 2001, 18, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Sinaga, E.; Jois, S.D.; Avery, M.; Makagiansar, I.T.; Tambunan, U.S.; Audus, K.L.; Siahaan, T.J. Increasing paracellular porosity by e-cadherin peptides: Discovery of bulge and groove regions in the EC1-domain of E-cadherin. Pharm Res. 2002, 19, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Ulapane, K.R.; On, N.; Kiptoo, P.; Williams, T.D.; Miller, D.W.; Siahaan, T.J. Improving brain delivery of biomolecules via BBB modulation in mouse and rat: Detection using MRI, NRIF, and mass spectrometry. Nanotheranostics 2017, 1, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Laksitorini, M.D.; Kiptoo, P.K.; On, N.H.; Thliveris, J.A.; Miller, D.W.; Siahaan, T.J. Modulation of intercellular junctions by cyclic-ADT peptides as a method to reversibly increase blood-brain barrier permeability. J. Pharm. Sci. 2015, 104, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Kiptoo, P.; Sinaga, E.; Calcagno, A.M.; Zhao, H.; Kobayashi, N.; Tambunan, U.S.; Siahaan, T.J. Enhancement of drug absorption through the blood-brain barrier and inhibition of intercellular tight junction resealing by E-cadherin peptides. Mol. Pharm 2011, 8, 239–249. [Google Scholar] [CrossRef] [PubMed]
- On, N.H.; Kiptoo, P.; Siahaan, T.J.; Miller, D.W. Modulation of blood-brain barrier permeability in mice using synthetic e-cadherin peptide. Mol. Pharm. 2014, 11, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Iorio, A.L.; da Ros, M.; Genitori, L.; Lucchesi, M.; Colelli, F.; Signorino, G.; Cardile, F.; Laffi, G.; de Martino, M.; Pisano, C.; et al. Tumor response of temozolomide in combination with morphine in a xenograft model of human glioblastoma. Oncotarget 2017, 8, 89595–89606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veringa, S.J.; Biesmans, D.; van Vuurden, D.G.; Jansen, M.H.; Wedekind, L.E.; Horsman, I.; Wesseling, P.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J.; et al. In vitro drug response and efflux transporters associated with drug resistance in pediatric high grade glioma and diffuse intrinsic pontine glioma. PLoS ONE 2013, 8, e61512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Ros, M.; Iorio, A.L.; Consolante, D.; Cardile, F.; Muratori, M.; Fantappiè, O.; Lucchesi, M.; Guidi, M.; Pisano, C.; Sardi, I. Morphine modulates doxorubicin uptake and improves efficacy of chemotherapy in an intracranial xenograft model of human glioblastoma. Am. J. Cancer Res. 2016, 6, 639–648. [Google Scholar] [PubMed]
- Sardi, I.; la Marca, G.; Cardellicchio, S.; Giunti, L.; Malvagia, S.; Genitori, L.; Massimino, M.; de Martino, M.; Giovannini, M.G. Pharmacological modulation of blood-brain barrier increases permeability of doxorubicin into the rat brain. Am. J. Cancer Res. 2013, 3, 424–432. [Google Scholar] [PubMed]
- Kim, S.; Jo, S.; Lee, H.; Kim, T.U.; Kim, I.C.; Yim, J.H.; Chung, H. Lobarstin enhances chemosensitivity in human glioblastoma T98G cells. Anticancer Res. 2013, 33, 5445–5451. [Google Scholar] [PubMed]
- McConnell, D.D.; McGreevy, J.W.; Williams, M.N.; Litofsky, N.S. Do Anti-Oxidants Vitamin D3, Melatonin, and α-Lipoic Acid Have Synergistic Effects with Temozolomide on Cultured Glioblastoma Cells? Medicines 2018, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [PubMed]
- Carman, A.J.; Mills, J.H.; Krenz, A.; Kim, D.G.; Bynoe, M.S. Adenosine receptor signaling modulates permeability of the blood-brain barrier. J. Neurosci. 2011, 31, 13272–13280. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Anders, N.M.; Mangraviti, A.; Wanjiku, T.M.; Sankey, E.W.; Liu, A.; Brem, H.; Tyler, B.; Rudek, M.A.; Grossman, S.A. The effect of regadenoson-induced transient disruption of the blood-brain barrier on temozolomide delivery to normal rat brain. J. Neurooncol. 2016, 126, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Facchino, S.; Abdouh, M.; Bernier, G. Brain cancer stem cells: Current status on glioblastoma multiforme. Cancers (Basel) 2011, 3, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, M.D.; Fosmark, S.; Hellwege, S.; Beier, D.; Kristensen, B.W.; Beier, C.P. Chemoresistance and chemotherapy targeting stem-like cells in malignant glioma. Adv. Exp. Med. Biol. 2015, 853, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Salmaggi, A.; Boiardi, A.; Gelati, M.; Russo, A.; Calatozzolo, C.; Ciusani, E.; Sciacca, F.L.; Ottolina, A.; Parati, E.A.; La Porta, C.; et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia 2006, 54, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Caldera, V.; Mellai, M.; Annovazzi, L.; Monzeglio, O.; Piazzi, A.; Schiffer, D. Mgmt hypermethylation and mdr system in glioblastoma cancer stem cells. Cancer Genom. Proteom. 2012, 9, 171–178. [Google Scholar]
- Squatrito, M.; Holland, E.C. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011, 71, 5945–5949. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. Dna repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Schmalz, P.G.; Shen, M.J.; Park, J.K. Treatment resistance mechanisms of malignant glioma tumor stem cells. Cancers (Basel) 2011, 3, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Pinnell, N.; Wen, P.Y.; D’Andrea, A. Targeting DNA repair and the cell cycle in glioblastoma. J. Neurooncol. 2012, 107, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Efferth, T.; Remacle, J. Chemotherapy-induced resistance by atp-binding cassette transporter genes. Biochim. Biophys. Acta 2007, 1775, 237–262. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, A.A.; Stromskaya, T.P. Transport proteins of the abc family and multidrug resistance of tumor cells. Biochemistry (Moscow) 2008, 73, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Bark, H.; Choi, C.H. Psc833, cyclosporine analogue, downregulates mdr1 expression by activating JNK/c-Jun/AP-1 and suppressing NF-κB. Cancer Chemother. Pharmacol. 2010, 65, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Zhou, S.; Wang, Y.; Liu, X.; Zhou, L.; Yin, P.; Fan, Z.; Li, Q. Cox-2 contributes to P-glycoprotein-mediated multidrug resistance via phosphorylation of c-JUN at ser63/73 in colorectal cancer. Carcinogenesis 2011, 32, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Barancík, M.; Bohácová, V.; Kvackajová, J.; Hudecová, S.; Krizanová, O.; Breier, A. Sb203580, a specific inhibitor of p38-MAKP pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. Eur. J. Pharm. Sci. 2001, 14, 29–36. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Han, Y.; Liu, J.; Zhang, J.; Li, F.; Wang, Y.; Liu, X.; Yao, L. Calebin-a induces apoptosis and modulates MAKP family activity in drug resistant human gastric cancer cells. Eur. J. Pharmacol. 2008, 591, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.W.; Wen, F. Tanshinone IIA acts via p38 MAKP to induce apoptosis and the down-regulation of ercc1 and lung-resistance protein in cisplatin-resistant ovarian cancer cells. Oncol. Rep. 2011, 25, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Rohlff, C.; Glazer, R.I. Regulation of multidrug resistance through the cAMP and EGF signalling pathways. Cell Signal. 1995, 7, 431–443. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.; Parsons, R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008, 27, 5477–5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, E.J.; Jin, C.; Kim, D.H. Activation of PI3K/AKT pathway by PTEN reduction and PIK3CA mrna amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol. Oncol. 2005, 97, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, N.; Saio, M.; Matsuo, A.; Sugiyama, Y.; Saji, S. The evaluation of gastric cancer sensitivity to 5-fu/cddp in terms of induction of apoptosis: Time- and p53 expression-dependency of anti-cancer drugs. Oncol. Rep. 2005, 14, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef] [PubMed]
- Zhukovskaya, N.; Rydberg, B.; Karran, P. Inactive o6-methylguanine-DNA methyltransferase in human cells. Nucleic Acids Res. 1992, 20, 6081–6090. [Google Scholar] [CrossRef] [PubMed]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for o6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. Mgmt promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. Mgmt gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene o6-methylguanine-dna methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797. [Google Scholar] [PubMed]
- Martinez, R.; Schackert, G.; Yaya-Tur, R.; Rojas-Marcos, I.; Herman, J.G.; Esteller, M. Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2007, 83, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. P53 effects both the duration of g2/m arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Bocangel, D.B.; Finkelstein, S.; Schold, S.C.; Bhakat, K.K.; Mitra, S.; Kokkinakis, D.M. Multifaceted resistance of gliomas to temozolomide. Clin. Cancer Res. 2002, 8, 2725–2734. [Google Scholar] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Visone, R.; Croce, C.M. mirnas and cancer. Am. J. Pathol. 2009, 174, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A MicroRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, J.; Yang, N.; Greshock, J.; Megraw, M.S.; Giannakakis, A.; Liang, S.; Naylor, T.L.; Barchetti, A.; Ward, M.R.; et al. MicroRNAs exhibit high frequency genomic alterations in human cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 9136–9141. [Google Scholar] [CrossRef] [PubMed]
- Bartels, C.L.; Tsongalis, G.J. MicroRNAs: Novel biomarkers for human cancer. Clin. Chem. 2009, 55, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xia, F.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, J.; Cui, Y.; Bian, X.; Bie, P.; et al. MicroRNA-122 sensitizes HCC cancer cells to adriamycin and vincristine through modulating expression of MDR and inducing cell cycle arrest. Cancer Lett. 2011, 310, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, N.; Yang, Z.; Zhou, B.; He, Y.; Weng, D.; Fang, Y.; Wu, P.; Chen, P.; Yang, X.; et al. MiR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J. Natl. Cancer Inst. 2013, 105, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Wang, Y.; Wang, C.X.; Yin, M.; Liu, H.L.; Chen, J.P.; Han, J.Q.; Wang, W.B. miRNA-155 mediates TAM resistance by modulating SOCS6-STAT3 signalling pathway in breast cancer. Am. J. Transl Res. 2015, 7, 2115–2126. [Google Scholar] [PubMed]
- Dong, Z.; Ren, L.; Lin, L.; Li, J.; Huang, Y. Effect of MicroRNA-21 on multidrug resistance reversal in A549/DDP human lung cancer cells. Mol. Med. Rep. 2015, 11, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841. [Google Scholar] [PubMed]
- Giunti, L.; da Ros, M.; Vinci, S.; Gelmini, S.; Iorio, A.L.; Buccoliero, A.M.; Cardellicchio, S.; Castiglione, F.; Genitori, L.; de Martino, M.; et al. Anti-miR21 oligonucleotide enhances chemosensitivity of t98g cell line to doxorubicin by inducing apoptosis. Am. J. Cancer Res. 2015, 5, 231–242. [Google Scholar] [PubMed]
- Bai, Y.; Liao, H.; Liu, T.; Zeng, X.; Xiao, F.; Luo, L.; Guo, H.; Guo, L. Mir-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (EAG1). Eur. J. Cancer 2013, 49, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Zhou, S.; Zhou, Z.; Yang, X.; Que, S.; Lan, J.; Qiu, Y.; Lin, Y. Mir-146b-5p suppresses glioblastoma cell resistance to temozolomide through targeting TRAF6. Oncol. Rep. 2017, 38, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Mingyi, M.; Qiu, X.; Qiu, Y. MicroRNA-101 reverses temozolomide resistance by inhibition of GSK3β in glioblastoma. Oncotarget 2016, 7, 79584–79595. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J. Clin. Oncol. 2007, 25, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Takeshima, H.; Kataoka, H. Anti-glioma therapy with temozolomide and status of the DNA-repair gene mgmt. Anticancer Res. 2009, 29, 4845–4854. [Google Scholar] [PubMed]
- Cai, S.; Xu, Y.; Cooper, R.J.; Ferkowicz, M.J.; Hartwell, J.R.; Pollok, K.E.; Kelley, M.R. Mitochondrial targeting of human o6-methylguanine DNA methyltransferase protects against cell killing by chemotherapeutic alkylating agents. Cancer Res. 2005, 65, 3319–3327. [Google Scholar] [CrossRef] [PubMed]
- Middlemas, D.S.; Stewart, C.F.; Kirstein, M.N.; Poquette, C.; Friedman, H.S.; Houghton, P.J.; Brent, T.P. Biochemical correlates of temozolomide sensitivity in pediatric solid tumor xenograft models. Clin. Cancer Res. 2000, 6, 998–1007. [Google Scholar] [PubMed]
- Silber, J.R.; Bobola, M.S.; Blank, A.; Schoeler, K.D.; Haroldson, P.D.; Huynh, M.B.; Kolstoe, D.D. The apurinic/apyrimidinic endonuclease activity of APE1/REF-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin. Cancer Res. 2002, 8, 3008–3018. [Google Scholar] [PubMed]
- Cheng, C.L.; Johnson, S.P.; Keir, S.T.; Quinn, J.A.; Ali-Osman, F.; Szabo, C.; Li, H.; Salzman, A.L.; Dolan, M.E.; Modrich, P.; et al. Poly(adp-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol. Cancer Ther. 2005, 4, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Ujifuku, K.; Mitsutake, N.; Takakura, S.; Matsuse, M.; Saenko, V.; Suzuki, K.; Hayashi, K.; Matsuo, T.; Kamada, K.; Nagata, I.; et al. MiR-195, miR-455-3p and miR-10a(*) are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010, 296, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Slaby, O.; Lakomy, R.; Fadrus, P.; Hrstka, R.; Kren, L.; Lzicarova, E.; Smrcka, M.; Svoboda, M.; Dolezalova, H.; Novakova, J.; et al. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma 2010, 57, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; Greco, S.J.; Rameshwar, P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015, 6, 1190–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.; Dong, L. Knockdown of MicroRNA-127 reverses adriamycin resistance via cell cycle arrest and apoptosis sensitization in adriamycin-resistant human glioma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6107–6116. [Google Scholar] [PubMed]
- Blower, P.E.; Chung, J.H.; Verducci, J.S.; Lin, S.; Park, J.K.; Dai, Z.; Liu, C.G.; Schmittgen, T.D.; Reinhold, W.C.; Croce, C.M.; et al. MicroRNAs modulate the chemosensitivity of tumor cells. Mol. Cancer Ther. 2008, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/PGP-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Dixon, J.E.; Durden, D.L.; Tonks, N.K.; Donner, D.B. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J. Biol. Chem. 2002, 277, 5484–5489. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; De Jesus, J.; et al. Oncogenic EGR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. MicroRNA-7 inhibits the epidermal growth factor receptor and the AKT pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef] [PubMed]
- Zocco, D.; Ferruzzi, P.; Cappello, F.; Kuo, W.P.; Fais, S. Extracellular vesicles as shuttles of tumor biomarkers and anti-tumor drugs. Front. Oncol. 2014, 4, 267. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Cerione, R.A. Microvesicles as mediators of intercellular communication in cancer. Methods Mol. Biol. 2014, 1165, 147–173. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma–Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Dehler, S.; Rushing, E.J.; Zaugg, K.; Hofer, S.; Yonekawa, Y.; Bertalanffy, H.; Valavanis, A.; Korol, D.; Rohrmann, S.; et al. Glioblastoma in the canton of Zurich, Switzerland revisited: 2005 to 2009. Cancer 2016, 122, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant iii peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Billingham, R.E.; Brent, L.; Medawar, P.B. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Billingham, R.E.; Brent, L.; Medawar, P.B.; Sparrow, E.M. Quantitative studies on tissue transplantation immunity. I. The survival times of skin homografts exchanged between members of different inbred strains of mice. Proc. R. Soc. Lond. Biol. Sci. 1954, 143, 43–58. [Google Scholar] [CrossRef]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.M.; Kochel, C.M.; Nirschl, C.J.; Durham, N.M.; Ruzevick, J.; Alme, A.; Francica, B.J.; Elias, J.; Daniels, A.; Dubensky, T.W.; et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin. Cancer Res. 2016, 22, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the cns in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jähnisch, H.; Füssel, S.; Kiessling, A.; Wehner, R.; Zastrow, S.; Bachmann, M.; Rieber, E.P.; Wirth, M.P.; Schmitz, M. Dendritic cell-based immunotherapy for prostate cancer. Clin. Dev. Immunol. 2010, 2010, 517493. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, L. Oncolytic viruses as immunotherapy: Progress and remaining challenges. Onco Targets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef] [PubMed]
- Jena, B.; Dotti, G.; Cooper, L.J. Redirecting t-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 2010, 116, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous dissemination of glioblastoma multiforme. Sci. Transl. Med. 2014, 6, 247ra101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, J.M.; Carter, B.S. Detection of glioblastoma in biofluids. J. Neurosurg. 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. https://doi.org/10.3390/ijms19102879
Da Ros M, De Gregorio V, Iorio AL, Giunti L, Guidi M, De Martino M, Genitori L, Sardi I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. International Journal of Molecular Sciences. 2018; 19(10):2879. https://doi.org/10.3390/ijms19102879
Chicago/Turabian StyleDa Ros, Martina, Veronica De Gregorio, Anna Lisa Iorio, Laura Giunti, Milena Guidi, Maurizio De Martino, Lorenzo Genitori, and Iacopo Sardi. 2018. "Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier" International Journal of Molecular Sciences 19, no. 10: 2879. https://doi.org/10.3390/ijms19102879
APA StyleDa Ros, M., De Gregorio, V., Iorio, A. L., Giunti, L., Guidi, M., De Martino, M., Genitori, L., & Sardi, I. (2018). Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. International Journal of Molecular Sciences, 19(10), 2879. https://doi.org/10.3390/ijms19102879