1. Introduction
Circular RNA (circRNA) is a class of important members of non-coding RNA family [
1], which is considered as abundant [
2], stable [
3], and ubiquitous in diverse eukaryotic organisms [
4,
5] and may present a high degree of conservatism across evolutions [
3,
6]. CircRNAs are those RNA molecules in which an upstream 5′ splice site covalently linked a downstream 3′ splice site as a circle (named as back-splicing) [
1,
7], and the closed-loop structure outperforms linear structure in resistant to RNA digestion [
4]. CircRNAs usually lack the poly(A) tails and reveal complicated structures which contains one or multiple exons, as well as introns, intergenic, and UTR regions [
7,
8,
9]. Benefiting from RNA-Seq technology, more and more circRNAs have been identified and validated, and accumulated studies reported and emphasized the importance of them in many physiological processes [
1,
4,
5], and associated to various complex diseases, e.g., digestive system cancers [
10], breast cancer [
11], diabetes [
12], etc. Some circRNAs were observed to be functional, such as suppressing the activity of specific microRNAs [
13], upregulating/downregulating the expressions of specific genes [
14]. Recently, a number of studies have shown the potential of circRNAs on multiple translation processes, which implies that some circRNAs may be translated to peptides or proteins in vivo [
15,
16]. Further explorations on circRNAs are strongly suggested, which will be of far-reaching significance especially on disease occurrence, development, and precision diagnosis and treatments [
17,
18,
19].
Detecting circRNAs is a basic, but crucial step for any related investigations [
20,
21]. In the experimental researches, RNase R is the popular protocol to enrich circRNAs [
22]. Along with the popularity of RNA-seq, it becomes the major way of identifying circRNAs from RNA-seq data by computational methods [
1,
20,
21,
23]. Many state-of-the-art algorithms have been proposed for circRNA detection, while several important comparison studies carefully discussed the advantages and disadvantages among different algorithmic strategies [
20,
21]. According to the requirement of gene annotation information, the existing approaches, varying in detecting and filtering methods, are classified as
de novo detection and annotation-dependent detection [
20,
21]. In addition, some data processing pipelines further introduce combined strategies to enhance the performance of circRNA detection [
23,
24]. In general, the annotation-dependent methods, such as CIRCexplorer [
25] or MapSplice [
26] are often more reliable in detecting the circRNAs that consist of annotated fragments but limit the usages on the species without complete annotation data. It also may suffer an accuracy loss when the circular junctions deviate the boundaries of exons. On the contrary, find_circ [
5], CIRI [
27], CIRI2 [
28] and other
de novo methods are able to handle more complicated cases [
7,
8,
9], where one of the computational challenges is balancing the sensitivity and precision of detecting and filtering the candidates [
21].
According to the current strategies on circRNA detecting and filtering, improving the accuracy of a detection algorithm is still an urgent need [
20,
21]. On the algorithmic level, the existing approaches could be improved in several aspects. Here we use CIRI2 as an example because it is suggested as one of the reputable algorithms and often outperforms in multiple comparison tests [
21,
27,
28]. CIRI2 first utilizes a paired chiastic clipping (PCC) signal filter based on the local re-alignment results provided by BWA–MEM. Then, it establishes a probabilistic model to estimate the possible origins of a set of sequential segments (
k-mers), each of which may have multiple mapping positions. This model not only improves the accuracy of detection but enhances the sensitivity as well. However, the PCC signals are often unreliable, either missed or incorrect, when the local regions for re-alignment involve complex junctions [
27,
28]. The flaws may originate from the following aspects: (1) BWA–MEM always selects the optimal alignment results in local re-alignment process, where some segments and their alignments may be ignored due to unbalanced segmentation. For example, it is possible that a back-spliced junction (BSJ) read has a much shorter segment flanking the junction comparing to the other one. However, these segments usually either cannot be mapped to the reference genome or are discarded by the mapper to avoid multiple “false-positive” alignments, both of which lead to the lack of PCC signals; (2) The shorter the segment that the algorithm cuts from a read, a higher the mapping error will be. The fortuitous matching may obscure the junction boundary between two separately mapped segments. In this case, the mapper often strapped in a deviation on compact idiosyncratic gapped alignment report (CIGAR) value. Thus, the insufficient PCC or CIGAR signals activate the filters or cross-validation filter, both of which lead to the lack of PCC signals as well; (3) Although the algorithm scans the unbalanced junction reads for the second round after clustering the circRNA candidates, the BSJ reads are not always able to provide paired signals due to the commonly low expression levels of circRNAs compared to the linear transcriptomes which consists of similar exons and/or introns, which present both 5′ splice site and 3′ splice site at the same time. This disables the algorithm and causes it to inadequately capture all of the BSJ reads.
From the above discussion, better incorporating the imperfect BSJ reads would intuitionally enhance the local data signals and further improve the performance of circRNA detection. Thus, in this article, we proposed an efficient algorithm for circRNA detection, implemented as CIRCPlus. CIRCPlus is a
de novo detection approach, which does not rely on the annotation information. CIRCPlus is supposed to be given a set of pair-ended reads with mapping results, which can be easily obtained by mapping tools, e.g., BWA–MEM, Bowtie2 [
29]. CIRCPlus incorporates the local multi-alignments (fragment similarities) between two sets of the BSJ reads spanning a candidate circular junction, which directly overcomes the dependence on PCC signals. Benefiting from this, the algorithm is further able to unbiasedly identify more subtypes of circRNAs, including (1) small circRNA, whose length is around or shorter than one insert-size; (2) complex circRNAs, which consists of complex circular sequences, e.g., involving one or multiple intron-retained fragments; and (3) a circRNA, which harbors one or more short exons around the junction. We conducted multiple groups of experiments to compare the proposed algorithm to the existing approaches when the simulation configurations alter. The experimental results demonstrate that CIRCPlus outperforms other
de novo approaches on both sensitivity and
F measurement in most of the cases, while CIRCPlus maintains a high level of precision as well. In addition, CIRCPlus held balanced sensitivity and reliability on the read datasets according to an objective assessment criteria based on RNase R-treated samples.
3. Materials and Methods
3.1. Framework of the Proposed Approach
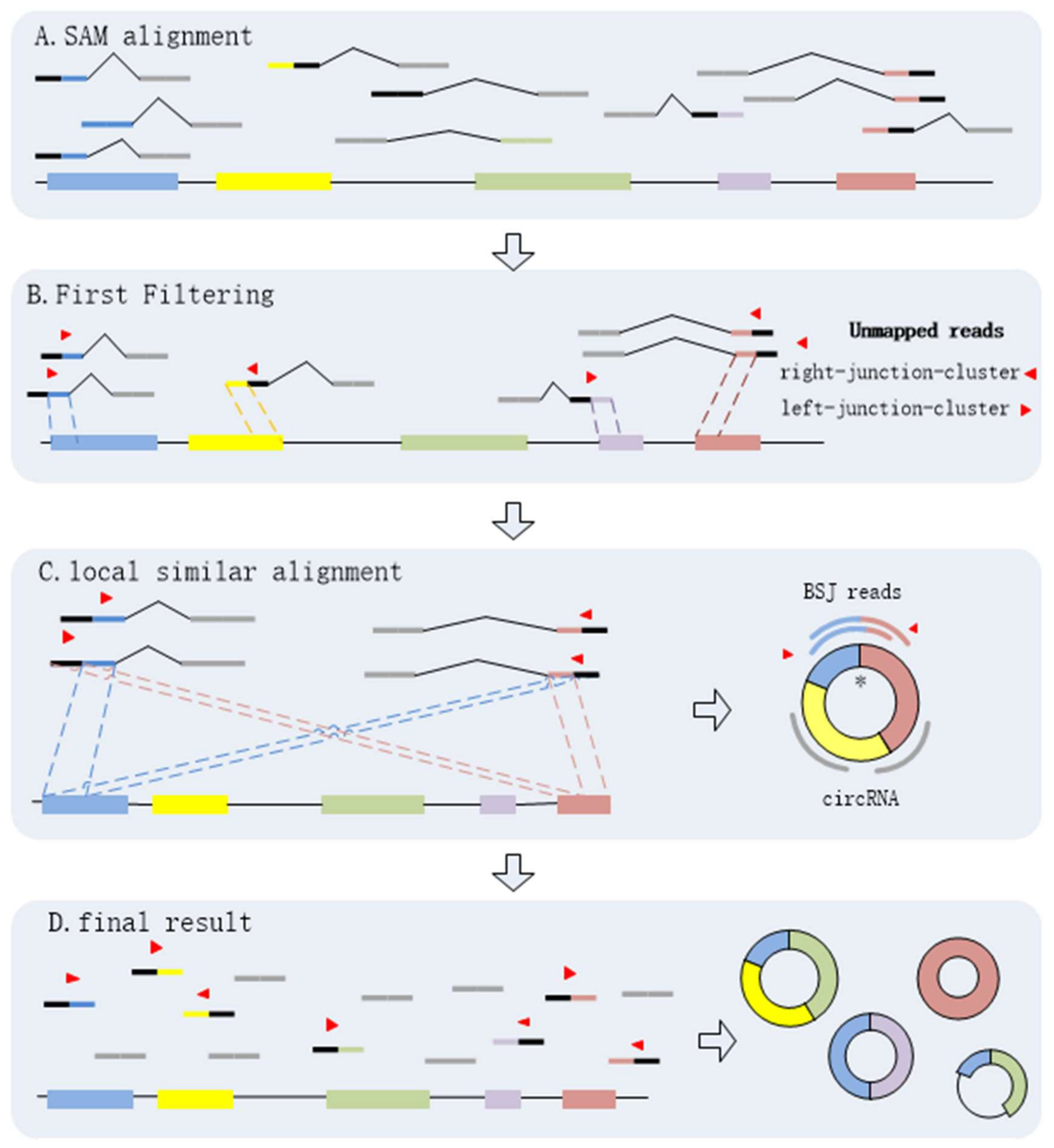
It is necessary to identify circRNAs at a high sensitivity with high precision regardless of circRNA expression levels and read alignment results. By analyzing the BSJ reads within a circRNA, we find that every BSJ read that spans the back-spliced site is comprised by two terminal sequences flanking the junctions. It is obvious that any two of BSJ reads must have local similar sequences. Here, we propose a novel algorithm, CIRCPlus, based on local sequence alignment between any two of BSJ reads and combined with a systematic filtering strategy to remove false positives, multithreading is implementation in CIRCplus to facilitate large dataset analysis. In CIRCPlus, the Sequence Alignment/Map (SAM) alignment file is generated by the BWA–MEM (the mapping tool for RNA-seq data which supports split mapping and calculates the junction information) and then used for identifying BSJ reads. The SAM file gives all alignment records of each read. CIRCPlus extracts all unmapped reads from the SAM file as the potential BSJ reads. These unmapped reads can be classified into two clusters according to alignment results from the SAM file. One cluster, called left-junction-cluster, contains the unmapped reads where the unmatched parts are on the left side, while the other, called right-junction-cluster, contains the unmapped reads where the unmatched parts are on the right side. Since a candidate BSJ read is considered to indicate a circRNA only when the mapping position of its paired read locates in the putative circRNA region on the reference genome, the unmapped reads in the two clusters can be filtered based on PEM (Paired-End Mapping) signals to refine the candidate BSJ reads. To identify the putative BSJ reads, considering that any pair of BSJ reads mapped in the same circular junction should both have a coverage region flanking 5′ and 3′ splice sites, and have an overlapped region termed as “local similar sequence”, the candidate BSJ reads are further filtered based on local similar sequences to obtain the putative BSJ reads. Here, a dynamic programming algorithm is employed to determine whether two reads from two clusters have local similar sequence, and outputs local similar sequence of these two reads. Finally, all of the putative BSJ reads are clustered to make the putative BSJ reads from the same circRNA in the same class.
The process of CIRCPlus can be summarized as following four steps:
- (1)
Extracting and classifying the unmapped reads;
- (2)
Filtering the unmapped reads based on PEM signal to get the candidate BSJ reads;
- (3)
Identifying the putative BSJ reads based on local similar sequences;
- (4)
Clustering all putative BSJ reads from the same circRNA.
Through the above steps, CIRCPlus is implemented to identify and characterize circRNAs. After the first and second steps, the candidate BSJ reads including the junction reads with PCC signals as well as unbalanced junction reads are obtained. Unlike the existing methods, e.g., CIRI, which extract a fixed size of anchors from the unmapped reads to identify potential backspliced junctions, CIRCPlus ignores such limitations for detecting unbalanced junction reads, and thus more candidate junction reads can be obtained for the next step. In the third step, CIRCPlus utilizes the local alignment to obtain the whole BSJ reads whether it has PCC signals or not, and the local alignment is useful to different read length during detection. As a result, the sensitivity of detection has been improved. The overall workflow of CIRCplus is shown in
Figure 4, each step will be described in detail separately.
3.2. Extracting and Classifying Unmapped Reads
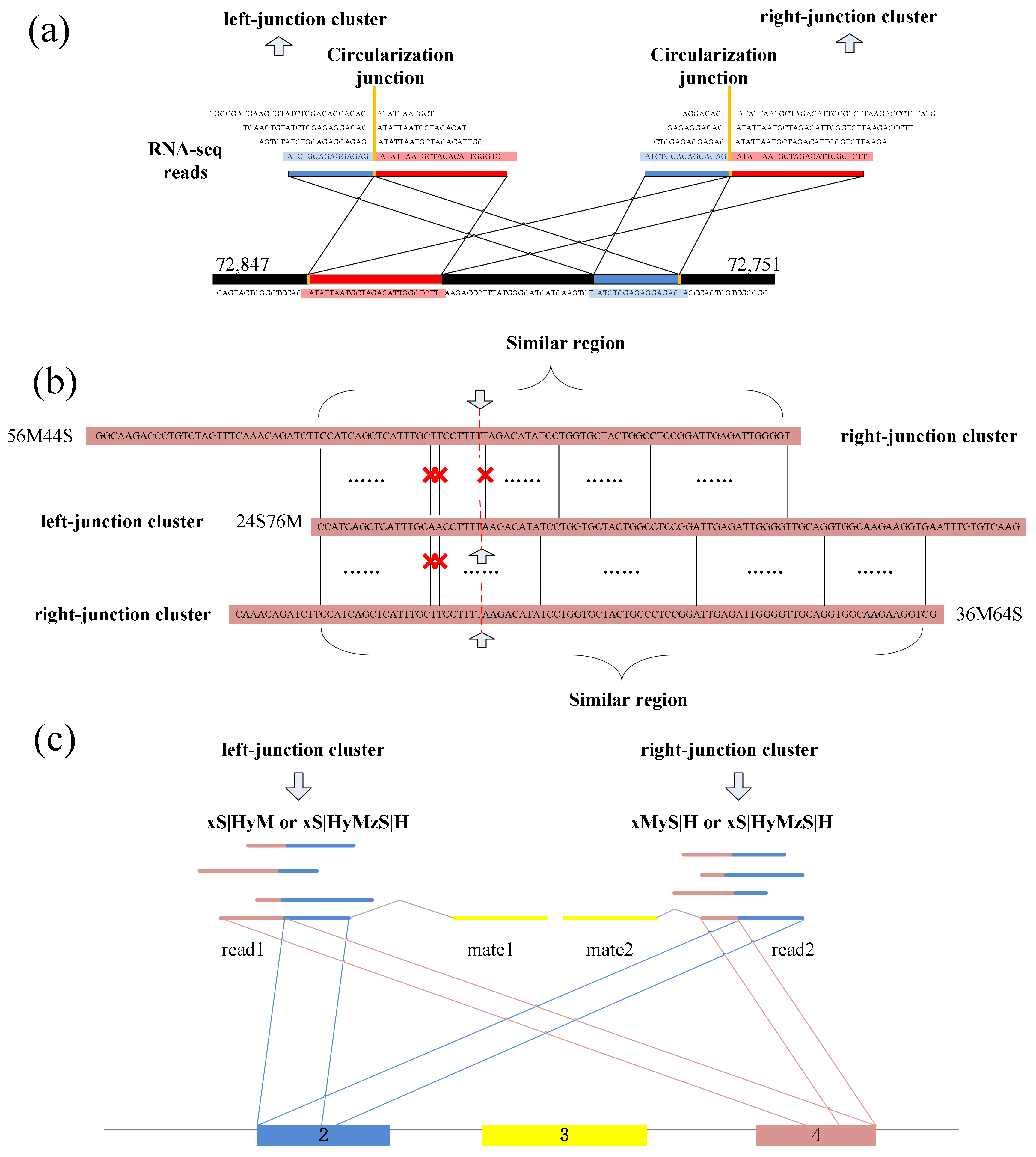
CIRCPlus requires the SAM alignment file generated by BWA–MEM. BWA–MEM aligns reads to the reference genome by mapping approaches and outputs to the confident alignment results to SAM file. During read mapping, some BSJ reads are mapped in 5′ splice site while some are mapped in 3′ splice site. Notably, some BSJ reads will have a pair of above alignment results, which named “paired chiastic clipping signals” (PCC signals) in CIRI. A typical junction is separately mapped with the reference in a corresponding two-segment style, as shown in
Figure 5a. Here, CIRCPlus extracts the unmapped reads which have corresponding alignment results and classifies the reads with CIGAR value in the form of xS|HyM in left-junction cluster (suppose that locate at 5′ splice site), while the reads with CIGAR value in the form of xMyS|H are put in right-junction cluster (suppose that locate at 3′ splice site).
In addition to the typical junction reads which are mapped to the reference genome in a two-segment style, some circRNAs have complex alignment features. In one case, if the exon flanking the junction of a circRNA is shorter than the read length, then some junction reads of the circRNA may inconsecutively map to the reference in a three-segment style, where two segments map to two exons flanking the junction and the third segment maps to the proximal part of the exon adjacent to the short flanking exon contained in the circRNA (
Figure 5b). In another case, if a circRNA is smaller than the read length, then it may also align to the reference in another form of the three-segment style, where two terminal segments separately overlap the terminal parts of the area where the middle segment aligns (
Figure 5c). In both situations, CIGAR values present the alignment features of xS|HyMzS|H at 5′ splice site and 3′splice site, and those reads are then put into the left-junction cluster and right-junction cluster simultaneously.
For each paired-end read, two ends are considered respectively. That is, each end is clustered into left-junction cluster or right-junction cluster according to its corresponding alignment records. When the reads have multiple mapping, the corresponding alignment records are separately taken into account, because sequencing errors or fortuitously matching bases may obscure the junction boundary between two separately aligned segments. Therefore, this method considers not only the PCC signals and unbalanced junction reads, but also the alignment results which just mapped at one of the junction sites. It simplifies the complexity of the second scanning of SAM in the existing method, and the candidate set can be expanded no matter whether it outputs the suitable alignment results or not. Here, the unmapped reads are put into two clusters based on CIGAR values, where left-junction cluster represents the potential chimeric junction reads which may be located at 5′ splice site within a circRNA, while right-junction cluster represents the potential chimeric junction reads which may be located at 3′ splice site.
3.3. Filtering Unmapped Reads
Because the two segments of a bona fide junction read represent the boundary of a circular RNA, the candidate junction read is considered to indicate a circRNA only when its paired read mapping position is within the putative circRNA region on the reference genome (
Figure 5d). Therefore, the unmapped reads in left-junction or right-junction cluster can be further filtered based on PEM signal to remove false positives.
For the junction reads in the left-junction cluster, the mapping positions represent the possible left boundaries of the circRNA, so the paired read should fall within the downstream area (5’ to 3’ direction) of its junction reads in the reference genome. Furthermore, for the reads in right-junction cluster, the mapping positions represent the possible right boundaries of the circRNA, so the paired read should fall within the upstream area (3’ to 5’ direction) of the junction reads. Here, the read direction is used to determine whether it is supported by the PEM signals. On one hand, the junction reads in the left-junction cluster should be the positive direction, while the paired read should be the negative direction. On the other hand, the junction reads in the right-junction cluster should be the negative direction, while the paired reads should be the positive direction. As a consequence, the paired-end reads with the same direction are filtered.
3.4. Identifying Putative BSJ Reads
Spanning the circular junction reads, termed as “BSJ reads”, are spliced by sequences flanking the junction. Those reads have different segment lengths flanking 5′ splice site and 3′ splice site respectively. As a consequence, a similarity sequence must exist between any two of BSJ reads within a circRNA, as shown in
Figure 5e. Therefore, a pair candidate BSJ reads mapped in the same circular junction should both have a coverage region flanking 5′ and 3′ splice sites. Local alignment, for determining similar regions between two reads, can be used to judge whether it outputs the similar sequence or not and then check the similar sequences if it spans the circular junction, as shown in
Figure 6a.
In the local alignment, the two reads for alignment should satisfy the following restrictions:
- (1)
The read from left-junction cluster should be located at the upstream of the read from right-junction cluster, and mapping distance between them along the genome reference should be reasonable.
- (2)
According to the PEM signals, their paired reads should fall within the region indicated by both junction reads.
- (3)
The two reads should be aligned to the same chromosome.
- (4)
The two reads should have the reasonable mapping scores.
For each pair of reads satisfying the above requirements, these two reads are compared to obtain their similar sequence. If a pair of reads exists similar sequences across the circular junction, these two reads are further detected. Here we suggest to use multithreading to analyze all alignment results between any supported pair of junction reads.
A dynamic programming algorithm is implemented to find the optimal local alignment with respect to the scoring system (which includes the substitution matrix and the gap-scoring scheme). Because the sequencing errors may lead to mismatch in the local sequence alignment between two BSJ reads, the number of mismatches is uncertain. So, a threshold for the numbers of mismatches and dashes is preset to filter the similar sequences, which makes the results better for detection. The threshold can be adjusted by user, as different read length may use different mismatch thresholds to obtain better precision.
Figure 6b shows an acceptable example during the detection.
3.5. Clustering All Putative BSJ Reads Within A CircRNA
Numerous of pairs of BSJ reads now have been generated though the previous steps, and for any pair, its results contain a read mapped to 5′ splice site and a read mapped to 3′ splice site within a circRNA. These BSJ reads are clustered subsequently according to their junction loci. Though fortuitously matching bases may obscure the junction boundary, the nearby junction loci can also be employed to cluster all circRNA junction reads, as shown in
Figure 6c.
Due to the split alignment strategy of BWA–MEM, the short splice read may contain false positive BSJ reads. To prevent false predictions resulting from fortuitously alignment results, we consider the mapping quality and the number of supporting BSJ reads to filter the false positive. After investigation of all reads, CIRCPlus summaries the mapping positions of all detected candidate BSJ reads. By comparing the read counts and the mapping quality of the reads, CIRCPlus further determines whether these reads reliably reflect a circRNA junction and whether the candidate circRNA should be kept until the final output or not.
4. Conclusions
In this article, we focused on the computational problem that identifying circRNAs from the next-generation RNA sequencing data. It is suggested that circRNAs with high abundance may associate with important functions. Detecting circRNAs directly from sequencing data is a computational challenge in bioinformatics. Several state-of-the-art methods are proposed and able to handle circRNA detections with different advantages and disadvantages, specifically on balancing the sensitivity and precision. We proposed a novel algorithm, CIRCPlus, to detect circRNAs from RNA-seq data. The proposed method directly computes the alignment results provided by BWA–MEM and implements an efficient de novo algorithm which simplifies the twice scanning strategy. CIRCPlus identifies the BSJ reads based on the local similar sequences, which was not considered in any of the existing methods. This strategy is able to identify more supporting BSJ reads of the circRNAs, where are usually ignored or misclassified by the existing methods. Benefiting from this, CIRCPlus is able to report circRNAs with high sensitivity and acceptable precision, and further obtain higher F1-score at the same time. Because the step “local alignment between two reads” is unaffected by read length, CIRCPlus also shows the better performance than the existing de novo methods on trimmed reads. The experimental results demonstrate that CIRCPlus is quite robust on both sensitivities and precisions varying the read depths, read lengths, the numbers of the preset circRNAs, as well as the different parameter settings on BWA-MEM, comparing to three popular methods CIRI2, CIRI, and find_circ in most of the simulation configurations. It is also tested on the paired HEK293 datasets, where CIRCPlus outperformed on both high sensitivity and low FDR. According to our limited knowledge on circRNA detection methods, we believe that CIRCPlus provides an efficient and unbiased circRNA detection tool for future circRNA studies. In the next study, we will further concentrate on increasing the precision in our algorithm.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}