Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cardiovascular Parameters
2.2. Hydroxyproline Concentration and Content in the Soluble and Insoluble Collagen and Total Hydroxyproline
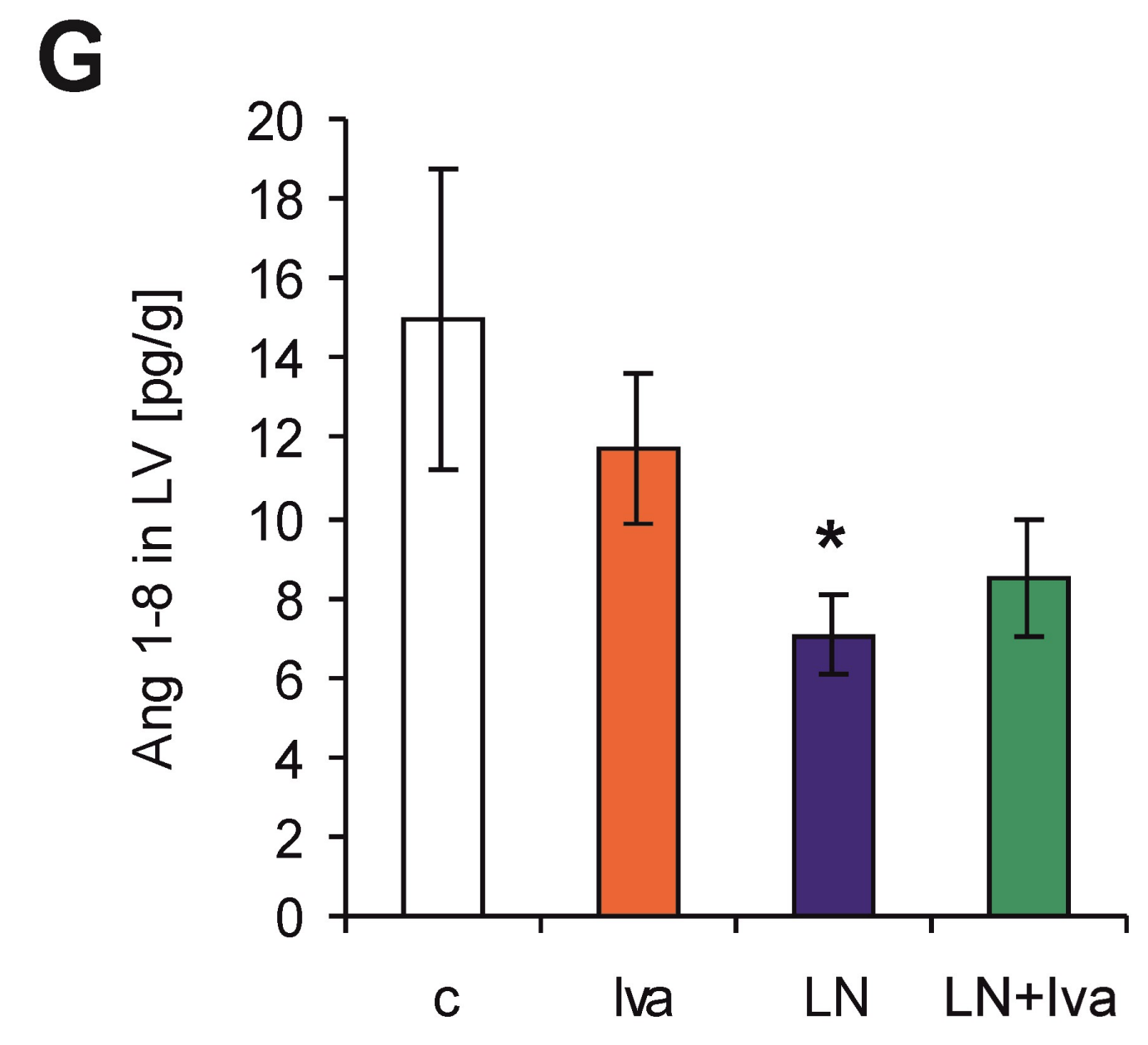
2.3. The Serum Concentration of Angiotensins and Aldosterone, Concentration of Angiotensin in the LV and Renin Activity (Measured and Surrogate)
2.4. Echocardiography
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Determination of Hydroxyproline
4.3. The Serum Concentration of Angiotensins and Aldosterone, the Concentration of Angiotensin in the LV and Renin Activity (Measured and Surrogate)
4.4. Echocardiography
4.5. Statistical Analysis
5. Limitations
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HR | Heart rate |
| LV | Left ventricle |
| L-NAME | NG-nitro-L-arginine methyl ester |
| Ang | Angiotensin |
| (AA2)-ratio | Aldosterone/angiotensin II ratio |
| HF | Heart failure |
| LVH | Left ventricular hypertrophy |
| Iva | Ivabradine group |
| LN | NG-nitro-L-arginine methyl ester group |
| c | Control group |
| LN+Iva | NG-nitro-L-arginine methyl ester plus ivabradine group |
| BW | Body weight |
| LVW | Left ventricular weight |
| RVW | Right ventricular weight |
| LVFS | Left ventricular fractional shortening |
| LVEF | Left ventricular ejection fraction |
| E velocity | Diastolic transmitral peak early filling velocity |
| A velocity | Diastolic transmitral peak late filling velocity |
| If | Funny current |
| RAAS | Renin-angiotensin-aldosterone system |
| SBP | Systolic blood pressure |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
References
- Silva, F.C.; Paiva, F.A.; Müller-Ribeiro, F.C.; Caldeira, H.M.; Fontes, M.; de Menezes, R.C.; Casali, K.R.; Fortes, G.H.; Tobaldini, E.; Solbiati, M.; et al. Chronic Treatment with Ivabradine Does Not Affect Cardiovascular Autonomic Control in Rats. Front. Physiol. 2016, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Gedik, N.; Witting, P.; Freedman, B.; Klöcker, N.; Heusch, G. Pleiotropic, heart rate-independent cardioprotection by ivabradine. Br. J. Pharmacol. 2015, 172, 4380–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcher, T.; Bernhardt, P.; Vasic, D.; Bach, H.; Durst, R.; Rottbauer, W.; Walcher, D. Ivabradine reduces chemokine-induced CD4-positive lymphocyte migration. Mediat. Inflamm. 2010, 2010, 751313. [Google Scholar] [CrossRef] [PubMed]
- Custodis, F.; Baumhäkel, M.; Schlimmer, N.; List, F.; Gensch, C.; Böhm, M.; Laufs, U. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation 2008, 117, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Fard, S.S.; Abreu-Gonzalez, P.; Bosa-Ojeda, F.; Consuegra-Sanchez, L.; Jiménez-Sosa, A.; Grande, A.S.; Kaski, J.C. Randomised, double-blind, placebo-controlled trial of ivabradine in patients with acute coronary syndrome: Effects of the If current inhibitor ivabradine on reduction of inflammation markers in patients with acute coronary syndrome—RIVIERA trial study design and rationale. Cardiovasc. Drugs Ther. 2009, 23, 243–247. [Google Scholar] [PubMed]
- Kröller-Schön, S.; Schulz, E.; Wenzel, P.; Kleschyov, A.L.; Hortmann, M.; Torzewski, M.; Oelze, M.; Renné, T.; Daiber, A.; Münzel, T. Differential effects of heart rate reduction with ivabradine in two models of endothelial dysfunction and oxidative stress. Basic Res. Cardiol. 2011, 106, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Becher, P.M.; Lindner, D.; Miteva, K.; Savvatis, K.; Zietsch, C.; Schmack, B.; Van Linthout, S.; Westermann, D.; Schultheiss, H.P.; Tschöpe, C. Role of heart rate reduction in the prevention of experimental heart failure: Comparison between If-channel blockade and β-receptor blockade. Hypertension 2012, 59, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Albaladejo, P.; Carusi, A.; Apartian, A.; Lacolley, P.; Safar, M.E.; Benetos, A. Effect of chronic heart rate reduction with ivabradine on carotid and aortic structure and function in normotensive and hypertensive rats. J. Vasc. Res. 2003, 40, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Bernatova, I.; Pelouch, V.; Simko, F. Protein remodelling of the heart in NO-deficient hypertension: The effect of captopril. J. Mol. Cell. Cardiol. 1997, 29, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Bernatova, I.; Pechanova, O.; Simko, F. Effect of captopril in L-NAME-induced hypertension on the rat myocardium, aorta, brain and kidney. Exp. Physiol. 1999, 84, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Matuskova, J.; Luptak, I.; Krajcirovicova, K.; Kucharska, J.; Gvozdjakova, A.; Babal, P.; Pechanova, O. Effect of simvastatin on remodeling of the left ventricle and aorta in L-NAME-induced hypertension. Life Sci. 2004, 74, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Matuskova, J.; Luptak, I.; Pincikova, T.; Krajcirovicova, K.; Stvrtina, S.; Pomsar, J.; Pelouch, V.; Paulis, L.; Pechánová, O. Spironolactone differently influences remodeling of the left ventricle and aorta in L-NAME-induced hypertension. Physiol. Res. 2007, 56, S25–S32. [Google Scholar] [PubMed]
- Bernatova, I.; Pechanova, O.; Pelouch, V.; Simko, F. Regression of chronic L-NAME-treatment-induced left ventricular hypertrophy: Effect of captopril. J. Mol. Cell. Cardiol. 2000, 32, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednarova, K.; Adamcova, M.; Paulis, L. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J. Hypertens. Suppl. 2009, 27, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Celec, P.; Tothova, L.; Vrankova, S.; Balazova, L.; Zorad, S.; et al. Lactacystin-induced model of hypertension in rats: Effects of melatonin and captopril. Int. J. Mol. Sci. 2017, 18, 1612. [Google Scholar] [CrossRef] [PubMed]
- Hegedusova, N.; Ondicova, K.; Mikova, L.; Horvathova, L.; Tillinger, A.; Mravec, B. The effect of ivabradine administration on heart rate, blood pressure, and secretion of epinephrine, norepinephrine, and corticosterone during stress response in rats. Cardiol. Lett. 2014, 23, 403–409. [Google Scholar]
- Luong, L.; Duckles, H.; Schenkel, T.; Mahmoud, M.; Tremoleda, J.L.; Wylezinska-Arridge, M.; Ali, M.; Bowden, N.P.; Villa-Uriol, M.C.; van der Heiden, K.; et al. Heart rate reduction with ivabradine promotes shear stress-dependent anti-inflamatory mechanisms in arteries. Thromb. Haemost. 2016, 116, 181–190. [Google Scholar] [PubMed]
- Simko, F.; Simko, J. The potential role of nitric oxide in the hypertrophic growth of the left ventricle. Physiol. Res. 2000, 49, 37–46. [Google Scholar] [PubMed]
- Koniari, I.; Mavrilas, D.; Apostolakis, E.; Papadimitriou, E.; Papadaki, H.; Papalois, A.; Poimenidi, E.; Xanthopoulou, I.; Hahalis, G.; Alexopoulos, D. Inhibition of Atherosclerosis Progression, Intimal Hyperplasia, and Oxidative Stress by Simvastatin and Ivabradine May Reduce Thoracic Aorta’s Stiffness in Hypercholesterolemic Rabbits. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Custodis, F.; Fries, P.; Müller, A.; Stamm, C.; Grube, M.; Kroemer, H.K.; Böhm, M.; Laufs, U. Heart rate reduction by ivabradine improves aortic compliance in apolipoprotein E-deficient mice. J. Vasc. Res. 2012, 49, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Dedkov, E.I.; Zheng, W.; Christensen, L.P.; Weiss, R.M.; Mahlberg-Gaudin, F.; Tomanek, R.J. Preservation of coronary reserve by ivabradine-induced reduction in heart rate in infarcted rats is associated with decrease in perivascular collagen. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H590–H598. [Google Scholar] [CrossRef] [PubMed]
- Busseuil, D.; Shi, Y.; Mecteau, M.; Brand, G.; Gillis, M.A.; Thorin, E.; Asselin, C.; Roméo, P.; Leung, T.K.; Latour, J.G.; et al. Heart rate reduction by ivabradine reduces diastolic dysfunction and cardiac fibrosis. Cardiology 2010, 117, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Rienzo, M.; Melka, J.; Bizé, A.; Sambin, L.; Jozwiak, M.; Su, J.B.; Hittinger, L.; Berdeaux, A.; Ghaleh, B. Ivabradine improves left ventricular function during chronic hypertension in conscious pigs. Hypertension 2015, 65, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Cho, K.I.; Kim, S.M.; Kim, N.; Han, J.; Kim, J.Y.; Kim, I.J. Heart rate reduction with ivabradine prevents thyroid hormone-induced cardiac remodeling in rat. Heart Vessels 2013, 28, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Ciobotaru, V.; Heimburger, M.; Louedec, L.; Heymes, C.; Ventura-Clapier, R.; Bedossa, P.; Escoubet, B.; Michel, J.B.; Mercadier, J.J.; Logeart, D. Effect of long-term heart rate reduction by If current inhibition on pressure overload-induced heart failure in rats. J. Pharmacol. Exp. Ther. 2008, 324, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, C.S.; Owens, R.E.; Bolorunduro, O.B.; Jha, S.K. Ivabradine: A Review of Labeled and off-Label Uses. Am. J. Cardiovasc. Drugs 2016, 16, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Dias da Silva, V.J.; Tobaldini, E.; Rocchetti, M.; Wu, M.A.; Malfatto, G.; Montano, N.; Zaza, A. Modulation of sympathetic activity and heart rate variability by ivabradine. Cardiovasc. Res. 2015, 108, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simko, F.; Baka, T.; Krajcirovicova, K.; Repova, K.; Aziriova, S.; Zorad, S.; Poglitsch, M.; Adamcova, M.; Reiter, R.J.; Paulis, L. Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension. Molecules 2018, 23, 265. [Google Scholar] [CrossRef] [PubMed]
- Muldowney, J.A.; Davis, S.N.; Vaughan, D.E.; Brown, N.J. NO synthase inhibition increases aldosterone in humans. Hypertension 2004, 44, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, T.; Tsuruya, K.; Ikeda, H.; Toyonaga, J.; Yamada, S.; Noguchi, H.; Tokumoto, M.; Kitazono, T. Systemic Aldosterone, But Not Angiotensin II, Plays a Pivotal Role in the Pathogenesis of Renal Injury in Chronic Nitric Oxide-Deficient Male Rats. Endocrinology 2015, 156, 2657–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funder, J.W. Aldosterone and mineralocorticoid receptors-physiology and pathophysiology. Int. J. Mol. Sci. 2017, 18, 1032. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Brutsaert, D.L. New concepts in diastolic dysfunction and diastolic heart failure: Part II: Causal mechanisms and treatment. Circulation 2002, 105, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.M.; Monasky, M.M.; Gu, L.; Taglieri, D.M.; Patel, B.G.; Liu, H.; Wang, Q.; Greener, I.; Dudley, S.C., Jr.; Solaro, R.J. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J. Mol. Cell. Cardiol. 2013, 56, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberman, G.A.; Fan, T.H.; Liu, H.; Jiao, Z.; Xiao, H.D.; Lovelock, J.D.; Boulden, B.M.; Widder, J.; Fredd, S.; Bernstein, K.E.; et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 2010, 121, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Shahbaz, A.U.; Kamalov, G.; Zhao, W.; Zhao, T.; Johnson, P.L.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C.; Weber, K.T. Mitochondria-targeted cardioprotection in aldosteronism. J. Cardiovasc. Pharmacol. 2011, 57, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Oki, K.; Kajikawa, M.; Nakashima, A.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Effect of aldosterone-producing adenoma on endothelial function and Rho-associated kinase activity in patients with primary aldosteronism. Hypertension 2015, 65, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Rajkovicova, R.; Simko, F. New developments in the pharmacological treatment of hypertension: Dead-end or a glimmer at the horizon? Curr. Hypertens. Rep. 2015, 17, 557. [Google Scholar] [CrossRef] [PubMed]
- Mulrow, P.J. Angiotensin II and aldosterone regulation. Regul. Pept. 1999, 80, 27–32. [Google Scholar] [CrossRef]
- Rebuffat, P.; Malendowicz, L.K.; Nussdorfer, G.G.; Mazzocchi, G. Stimulation of endogenous nitric oxide production is involved in the inhibitory effect of adrenomedullin on aldosterone secretion in the rat. Peptides 2001, 22, 923–926. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Holmes, B.B.; McCoy, M.J.; Hillard, C.J.; Campbell, W.B. Chronic administration of nitric oxide reduces angiotensin II receptor type 1 expression and aldosterone synthesis in Zona glomerulosa cells. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E820–E827. [Google Scholar] [CrossRef] [PubMed]
- Pelouch, V.; Milerova, M.; Ostadal, B.; Samanek, M.; Hucin, B. Protein profiling of human atrial and ventricular musculature: The effect of normoxaemia and hypoxaemia in congenital heart diseases. Physiol. Res. 1993, 42, 235–242. [Google Scholar] [PubMed]
- Reddy, G.K.; Enwemeka, C.S. A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 1996, 29, 225–229. [Google Scholar] [CrossRef]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Domenig, O.; Manzel, A.; Grobe, N.; Königshausen, E.; Kaltenecker, C.C.; Kovarik, J.J.; Stegbauer, J.; Gurley, S.B.; van Oyen, D.; Antlanger, M.; et al. Mediator of Alternative Renin-Angiotensin-System Activation in the Murine and Human Kidney. Sci. Rep. 2016, 6, 33678. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Goliasch, G.; Wurm, R.; Novak, J.; Strunk, G.; Gyöngyösi, M.; Poglitsch, M.; Säemann, M.D.; Hülsmann, M. Low- and High-renin Heart Failure Phenotypes with Clinical Implications. Clin. Chem. 2018, 64, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Baka, T.; Hodosy, J.; Krajcirovicova, K.; Repova, K.; Aziriova, S.; Domonkos, E.; Borbelyova, V.; Slavkovsky, P.; Zorad, S.; Celec, P.; et al. 17β-Estradiol treatment reversed left ventricular dysfunction in castrated male rats: An echocardiographic study. Can. J. Physiol. Pharmacol. 2018, 96, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rigel, D.F. Echocardiographic examination in rats and mice. Methods Mol. Biol. 2009, 573, 139–155. [Google Scholar] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simko, F.; Baka, T.; Poglitsch, M.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Zorad, S.; Adamcova, M.; Paulis, L. Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. Int. J. Mol. Sci. 2018, 19, 3017. https://doi.org/10.3390/ijms19103017
Simko F, Baka T, Poglitsch M, Repova K, Aziriova S, Krajcirovicova K, Zorad S, Adamcova M, Paulis L. Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. International Journal of Molecular Sciences. 2018; 19(10):3017. https://doi.org/10.3390/ijms19103017
Chicago/Turabian StyleSimko, Fedor, Tomas Baka, Marko Poglitsch, Kristina Repova, Silvia Aziriova, Kristina Krajcirovicova, Stefan Zorad, Michaela Adamcova, and Ludovit Paulis. 2018. "Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension" International Journal of Molecular Sciences 19, no. 10: 3017. https://doi.org/10.3390/ijms19103017
APA StyleSimko, F., Baka, T., Poglitsch, M., Repova, K., Aziriova, S., Krajcirovicova, K., Zorad, S., Adamcova, M., & Paulis, L. (2018). Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. International Journal of Molecular Sciences, 19(10), 3017. https://doi.org/10.3390/ijms19103017