Movement of the RecG Motor Domain upon DNA Binding Is Required for Efficient Fork Reversal


Abstract
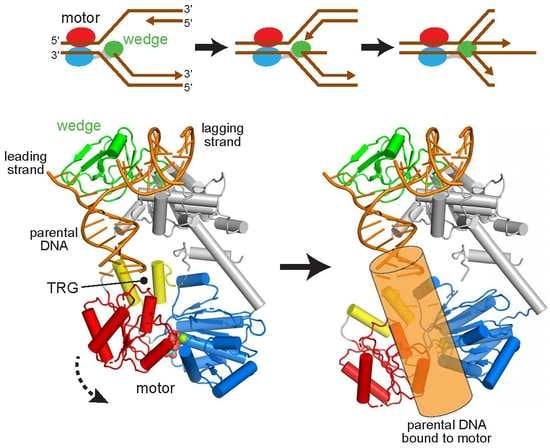
:
1. Introduction
2. Results
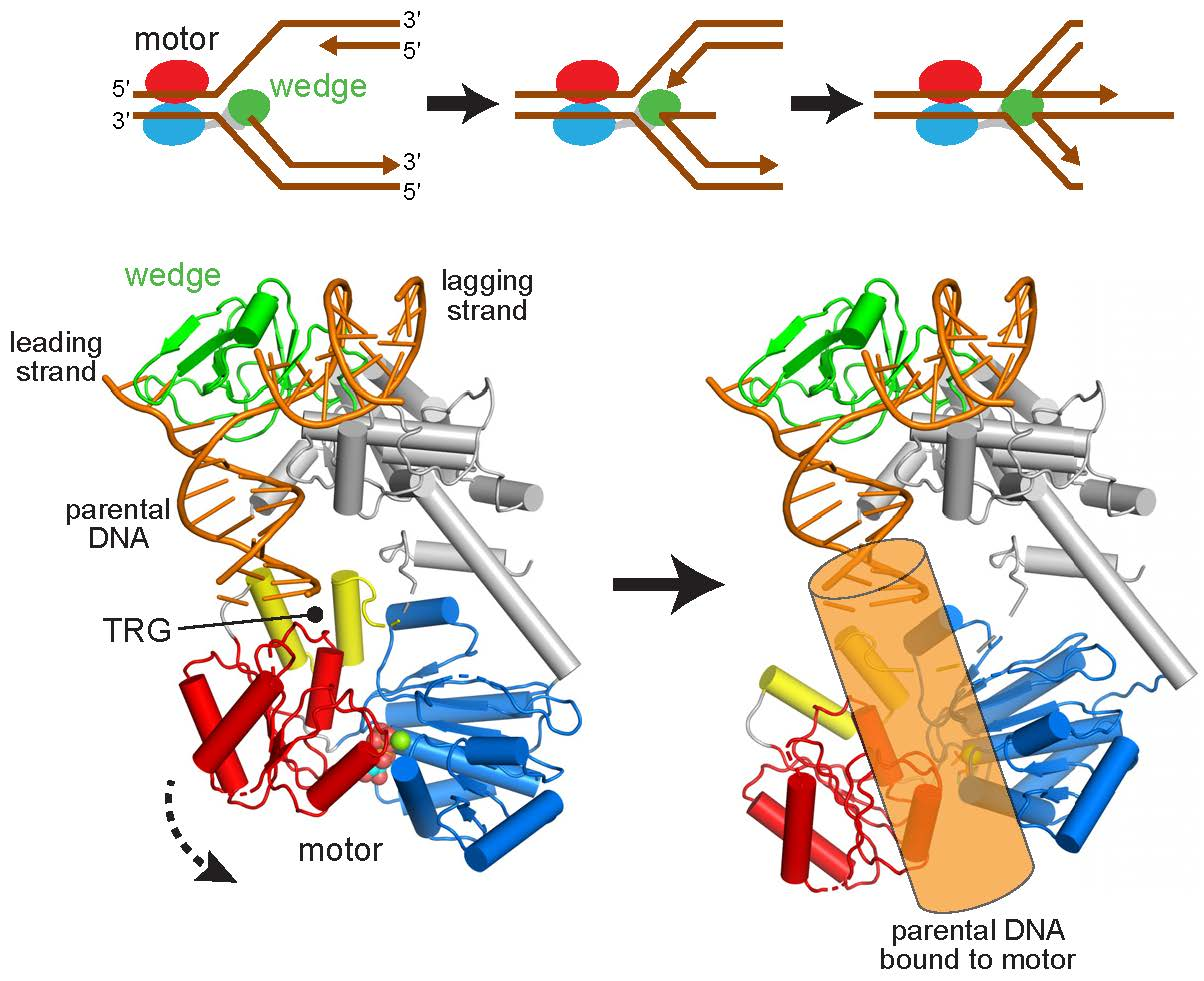
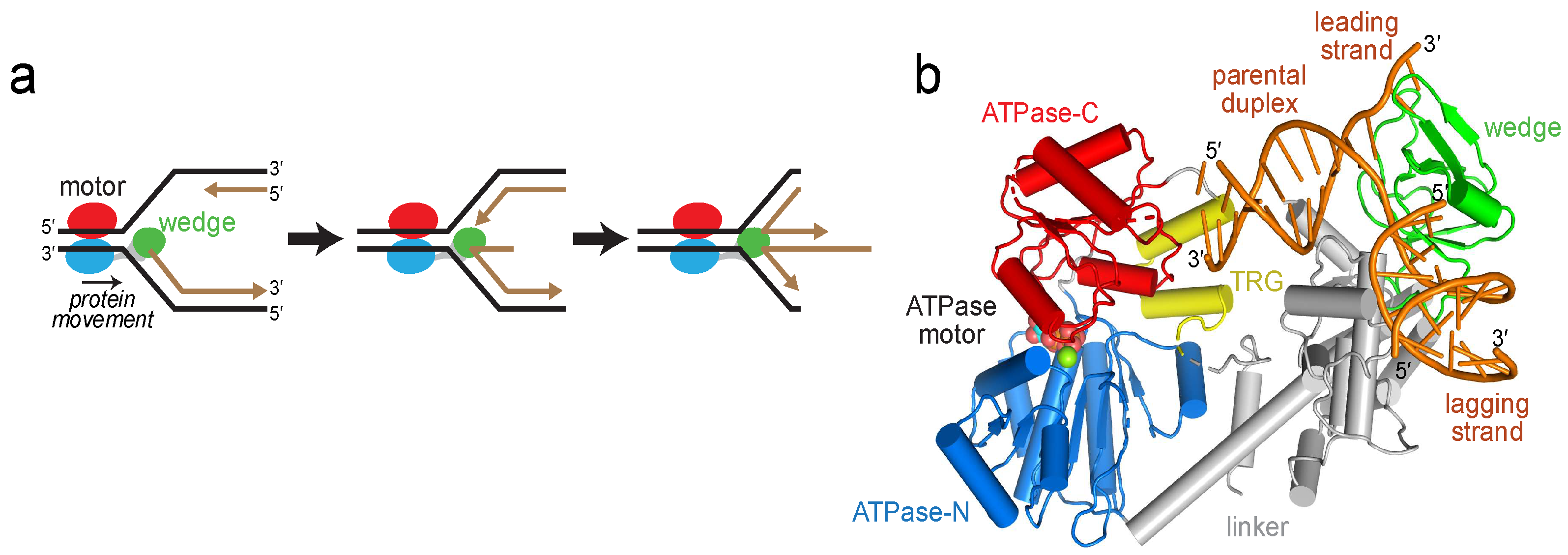
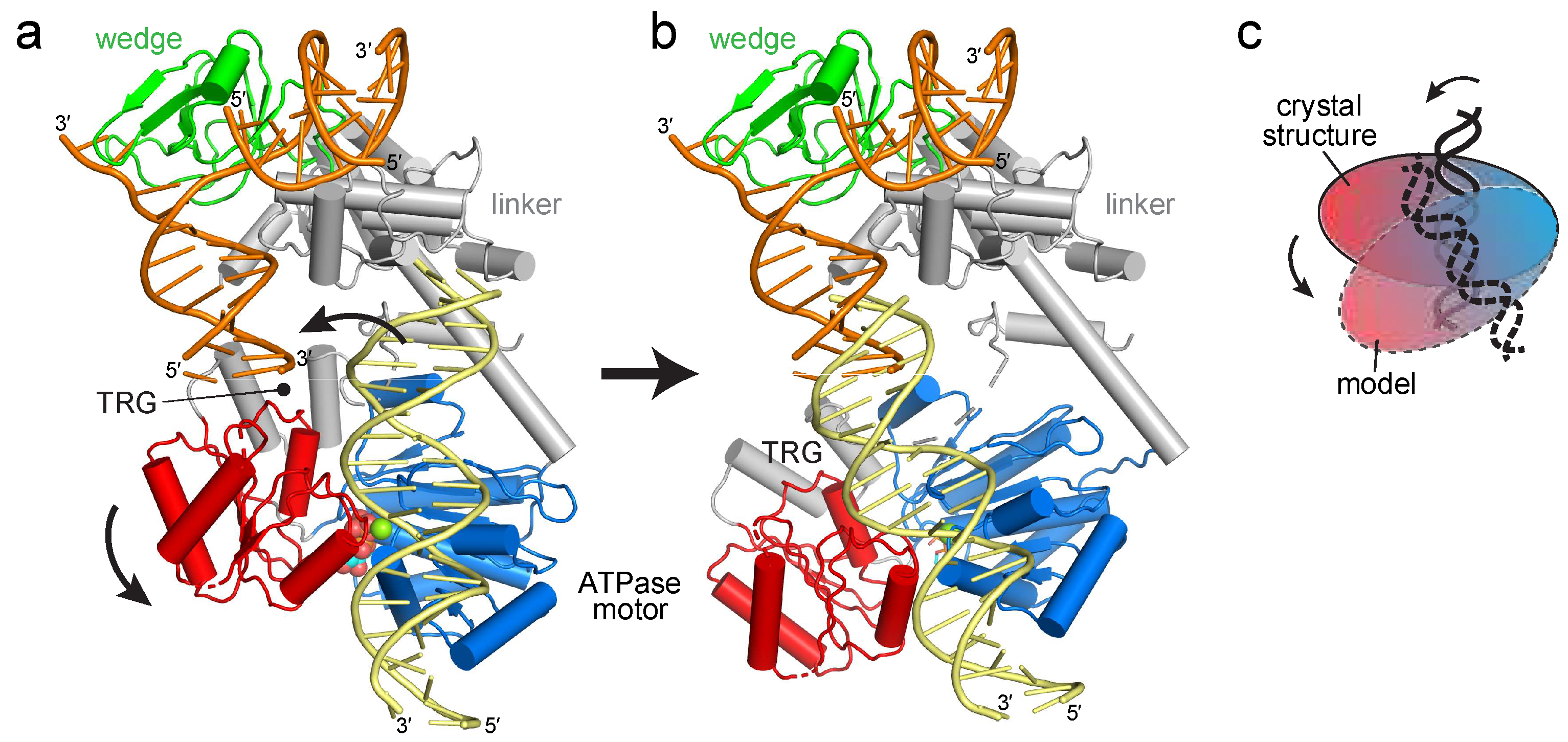
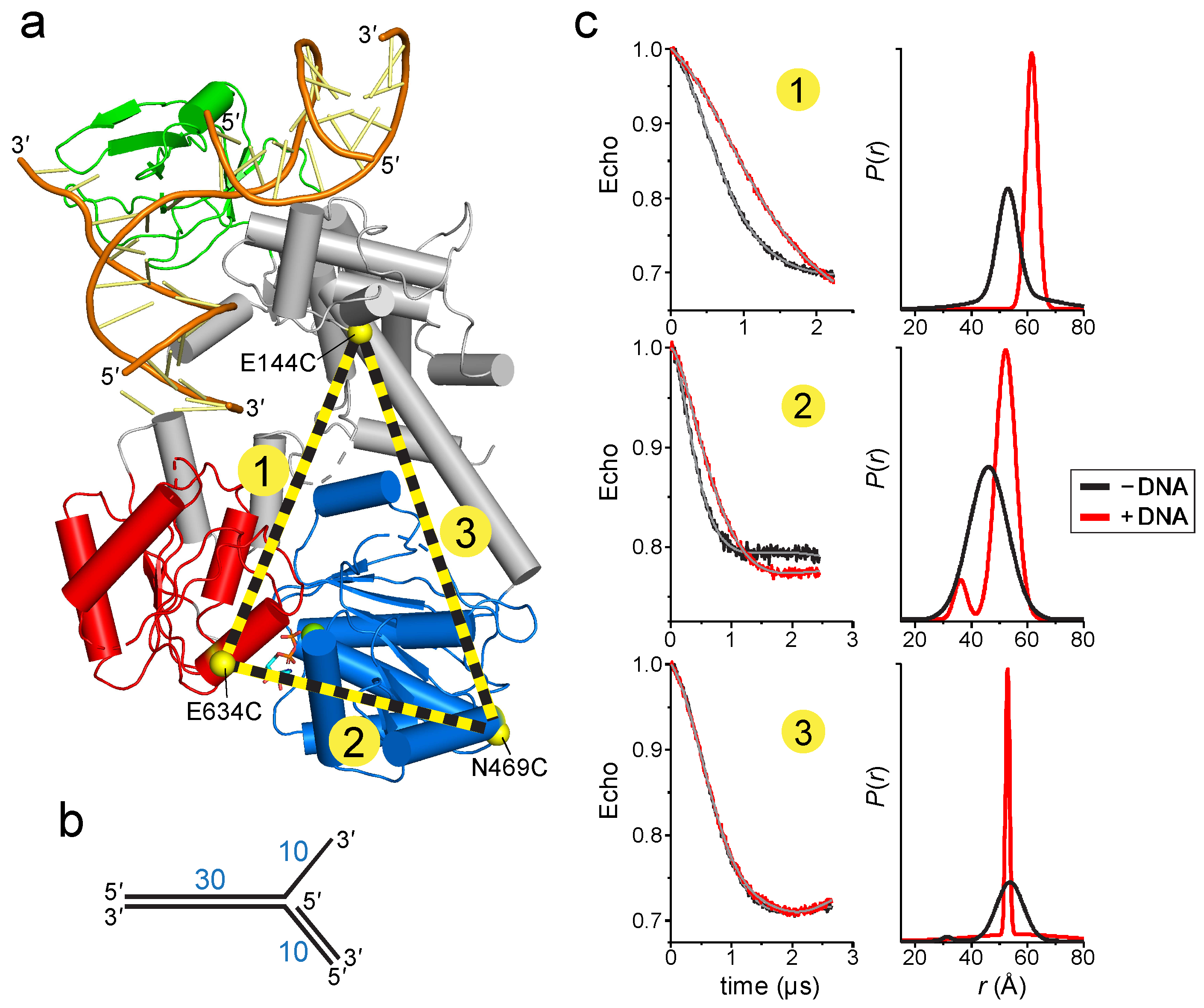
2.1. Reorientation of the RecG Motor Domain to Accommodate the Parental DNA Duplex
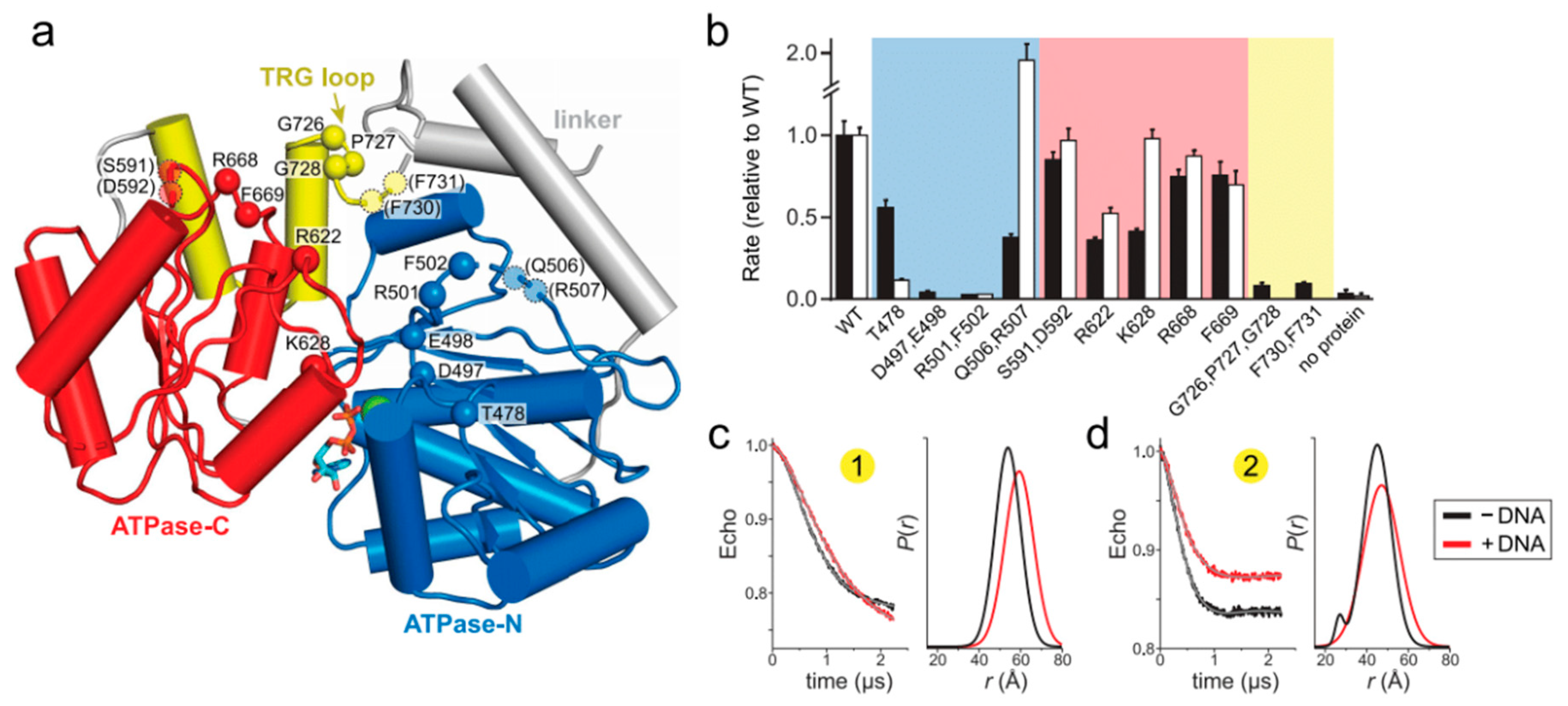
2.2. Mutation of the TRG Motif Attenuates RecG Conformational Changes upon DNA Binding
3. Discussion
4. Materials and Methods
4.1. Protein Purification
4.2. EPR
4.3. ATPase Assay
4.4. Fork Reversal Activity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine 5′-triphosphate |
| ATPase | adenosine triphosphatase |
| DEER | double electron-electron resonance |
| DTT | dithiothreitol |
| EDTA | ethylenediaminetetraacetic acid |
| EPR | electron paramagnetic resonance |
| MTSL | [1-oxy-2,2,5,5-tetramethyl-pyrolline-3-methyl]-methanethiosulfonate |
| NTA | nitrilotriacetic acid |
| SF2 | superfamily 2 |
| SRD | substrate recognition domain |
| SSC | saline-sodium citrate |
| TCEP | tris(2-carboxyethyl)phosphine |
| TRG | translocation in RecG |
References
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, D. Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst) 2015, 32, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marians, K.J. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Tatsumi, M. Replicative bypass repair of ultraviolet damage to DNA of mammalian cells: Caffeine sensitive and caffeine resistant mechanisms. Mutat. Res. 1976, 37, 91–110. [Google Scholar] [CrossRef]
- Higgins, N.P.; Kato, K.; Strauss, B. A model for replication repair in mammalian cells. J. Mol. Biol. 1976, 101, 417–425. [Google Scholar] [CrossRef]
- Atkinson, J.; McGlynn, P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009, 37, 3475–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.G.; Rudolph, C.J. 25 years on and no end in sight: A perspective on the role of RecG protein. Curr. Genet. 2016, 62, 827–840. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, P.; Lloyd, R.G. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002, 18, 413–419. [Google Scholar] [CrossRef]
- Bianco, P.R. I came to a fork in the DNA and there was RecG. Prog. Biophys. Mol. Biol. 2015, 117, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, R.G. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J. Bacteriol. 1991, 173, 5414–5418. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.G.; Buckman, C. Genetic analysis of the recG locus of Escherichia coli K-12 and of its role in recombination and DNA repair. J. Bacteriol. 1991, 173, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol. Microbiol. 2009, 74, 940–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.A.; Lloyd, R.G. Avoiding chromosome pathology when replication forks collide. Nature 2013, 500, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courcelle, J.; Hanawalt, P.C. RecA-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 2003, 37, 611–646. [Google Scholar] [CrossRef] [PubMed]
- Gregg, A.V.; McGlynn, P.; Jaktaji, R.P.; Lloyd, R.G. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell 2002, 9, 241–251. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Briggs, G.S.; Lloyd, R.G. Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 2010, 9, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Kowalczykowski, S.C. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000, 25, 156–165. [Google Scholar] [CrossRef]
- West, S.C. Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 1997, 31, 213–244. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.G.; Sharples, G.J. Dissociation of synthetic Holliday junctions by E. coli RecG protein. EMBO J. 1993, 12, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Whitby, M.C.; Ryder, L.; Lloyd, R.G. Reverse branch migration of Holliday junctions by RecG protein: A new mechanism for resolution of intermediates in recombination and DNA repair. Cell 1993, 75, 341–350. [Google Scholar] [CrossRef]
- Gupta, S.; Yeeles, J.T.; Marians, K.J. Regression of replication forks stalled by leading-strand template damage: I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the holliday junctions formed by RecG preferentially. J. Biol Chem 2014, 289, 28376–28387. [Google Scholar] [CrossRef] [PubMed]
- Azeroglu, B.; Mawer, J.S.; Cockram, C.A.; White, M.A.; Hasan, A.M.; Filatenkova, M.; Leach, D.R. RecG Directs DNA Synthesis during Double-Strand Break Repair. PLoS Genet. 2016, 12, e1005799. [Google Scholar] [CrossRef] [PubMed]
- Azeroglu, B.; Leach, D.R.F. RecG controls DNA amplification at double-strand breaks and arrested replication forks. FEBS Lett. 2017, 591, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley-Smith, S.L.; Dimude, J.U.; Taylor, T.; Forrester, N.M.; Upton, A.L.; Lloyd, R.G.; Rudolph, C.J. Chromosomal over-replication in Escherichia coli recG cells is triggered by replication fork fusion and amplified if replichore symmetry is disturbed. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fairman-Williams, M.E.; Guenther, U.P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Abd Wahab, S.; Choi, M.; Bianco, P.R. Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J. Biol. Chem. 2013, 288, 26397–26409. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, P.; Lloyd, R.G. Rescue of stalled replication forks by RecG: Simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. USA 2001, 98, 8227–8234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, M.R.; Scaife, S.; Wigley, D.B. Structural analysis of DNA replication fork reversal by RecG. Cell 2001, 107, 79–89. [Google Scholar] [CrossRef]
- Manosas, M.; Perumal, S.K.; Bianco, P.R.; Ritort, F.; Benkovic, S.J.; Croquette, V. RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat. Commun. 2013, 4, 2368. [Google Scholar] [CrossRef] [PubMed]
- Briggs, G.S.; Mahdi, A.A.; Wen, Q.; Lloyd, R.G. DNA binding by the substrate specificity (wedge) domain of RecG helicase suggests a role in processivity. J. Biol. Chem. 2005, 280, 13921–13927. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.A.; Briggs, G.S.; Sharples, G.J.; Wen, Q.; Lloyd, R.G. A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J. 2003, 22, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, A.L.; Smith, A.J.; Savery, N.J. A DNA translocation motif in the bacterial transcription--repair coupling factor, Mfd. Nucleic Acids Res. 2003, 31, 6409–6418. [Google Scholar] [CrossRef] [PubMed]
- Deaconescu, A.M.; Chambers, A.L.; Smith, A.J.; Nickels, B.E.; Hochschild, A.; Savery, N.J.; Darst, S.A. Structural basis for bacterial transcription-coupled DNA repair. Cell 2006, 124, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Marr, M.T.; Roberts, J.W. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 2002, 109, 757–767. [Google Scholar] [CrossRef]
- Deaconescu, A.M.; Savery, N.; Darst, S.A. The bacterial transcription repair coupling factor. Curr. Opin. Struct. Biol. 2007, 17, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savery, N.J. The molecular mechanism of transcription-coupled DNA repair. Trends Microbiol. 2007, 15, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Masai, H. Stabilization of a stalled replication fork by concerted actions of two helicases. J. Biol. Chem. 2006, 281, 3484–3493. [Google Scholar] [CrossRef] [PubMed]
- Ayala, R.; Willhoft, O.; Aramayo, R.J.; Wilkinson, M.; McCormack, E.A.; Ocloo, L.; Wigley, D.B.; Zhang, X. Structure and regulation of the human INO80-nucleosome complex. Nature 2018, 556, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Schall, K.; Kostrewa, D.; Lakomek, K.; Strauss, M.; Moldt, M.; Hopfner, K.P. Structural basis for ATP-dependent chromatin remodelling by the INO80 complex. Nature 2018, 556, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Xia, X.; Li, X.; Chen, Z. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 2017, 544, 440–445. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnung, L.; Vos, S.M.; Wigge, C.; Cramer, P. Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 2017, 550, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Durr, H.; Korner, C.; Muller, M.; Hickmann, V.; Hopfner, K.P. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell 2005, 121, 363–373. [Google Scholar] [CrossRef] [PubMed]
- McHaourab, H.S.; Steed, P.R.; Kazmier, K. Toward the fourth dimension of membrane protein structure: Insight into dynamics from spin-labeling EPR spectroscopy. Structure 2011, 19, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.M. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008, 37, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Zegeye, E.D.; Balasingham, S.V.; Laerdahl, J.K.; Homberset, H.; Kristiansen, P.E.; Tonjum, T. Effects of conserved residues and naturally occurring mutations on Mycobacterium tuberculosis RecG helicase activity. Microbiology 2014, 160, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Kim, J.L. Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 1999, 73, 8798–8807. [Google Scholar] [PubMed]
- Windgassen, T.A.; Keck, J.L. An aromatic-rich loop couples DNA binding and ATP hydrolysis in the PriA DNA helicase. Nucleic Acids Res. 2016, 44, 9745–9757. [Google Scholar] [CrossRef] [PubMed]
- Zittel, M.C.; Keck, J.L. Coupling DNA-binding and ATP hydrolysis in Escherichia coli RecQ: Role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 2005, 33, 6982–6991. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.A.; Cortez, D. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.C.; Rambo, R.P.; Greer, B.; Pritchett, M.; Tainer, J.A.; Cortez, D.; Eichman, B.F. A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 7618–7623. [Google Scholar] [CrossRef] [PubMed]
- Kile, A.C.; Chavez, D.A.; Bacal, J.; Eldirany, S.; Korzhnev, D.M.; Bezsonova, I.; Eichman, B.F.; Cimprich, K.A. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol. Cell. 2015, 58, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; Durr, H.; Hopfner, K.P.; Michaelis, J. Conformational changes of a Swi2/Snf2 ATPase during its mechano-chemical cycle. Nucleic Acids Res. 2008, 36, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Michaelis, J. Mechanisms of nucleic acid translocases: Lessons from structural biology and single-molecule biophysics. Curr. Opin. Struct. Biol. 2007, 17, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, L.; Tian, Y.; Xia, X.; Chen, Z. Structure and regulation of the chromatin remodeller ISWI. Nature 2016, 540, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.R.; Pottinger, S.; Tan, H.Y.; Nguyenduc, T.; Rex, K.; Varshney, U. The IDL of E. coli SSB links ssDNA and protein binding by mediating protein-protein interactions. Protein Sci. 2017, 26, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Betous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell. Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeschke, G. DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Verhalen, B.; Stein, R.A.; Wen, P.C.; Tajkhorshid, E.; McHaourab, H.S. Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. Elife 2014, 3, e02740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, R.A.; Beth, A.H.; Hustedt, E.J. A Straightforward Approach to the Analysis of Double Electron-Electron Resonance Data. Methods Enzymol. 2015, 563, 531–567. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EPR |
| F1—(32P)GGTCAGTCCTGTCTTCGGCAAAGCTCCATGATCATTGGCA |
| F2—CGCCGGGCCGCATGGAGCTTTGCCGAAGACAGGACTGACC |
| F3—CGGCCCGGCG |
| ATPase |
| J1—GGGTGAACCTGCAGGTGGGCCAGCTCCATGATCATTGGCAATCGTCAAGCTTTATGCCGT |
| J2—CGATGGACACGTCTTATGTGTGCAGTGCTCGCATGGAGCTGGCCCACCTGCAGGTTCACCC |
| J3—CATGTAGCGGCTGGCGTCTTAAAGATGTCCCGAGCACTGCACACATAAGACGTGTCCATCG |
| J4—ACGGCATAAAGCTTGACGATTGCCAATGATGGACATCTTTAAGACGCCAGCCGCTACATG |
| Fork Reversal 2 |
| F48—(32P)ACGCTGCCGAATTCTACCAGTGCCTTGCTAGGACATCTTTGCCCACCTGCAGGTTCACCC |
| F50—GGGTGAACCTGCAGGTGGGCAAAGATGTCC |
| F52—GGGTGAACCTGCAGGTGGGCAAAGATGTCCCAGCAAGGCACTGGTAGAATTCGGCAGCGTC |
| F53—GGACATCTTTGCCCACCTGCAGGTTCACCC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warren, G.M.; Stein, R.A.; Mchaourab, H.S.; Eichman, B.F. Movement of the RecG Motor Domain upon DNA Binding Is Required for Efficient Fork Reversal. Int. J. Mol. Sci. 2018, 19, 3049. https://doi.org/10.3390/ijms19103049
Warren GM, Stein RA, Mchaourab HS, Eichman BF. Movement of the RecG Motor Domain upon DNA Binding Is Required for Efficient Fork Reversal. International Journal of Molecular Sciences. 2018; 19(10):3049. https://doi.org/10.3390/ijms19103049
Chicago/Turabian StyleWarren, Garrett M., Richard A. Stein, Hassane S. Mchaourab, and Brandt F. Eichman. 2018. "Movement of the RecG Motor Domain upon DNA Binding Is Required for Efficient Fork Reversal" International Journal of Molecular Sciences 19, no. 10: 3049. https://doi.org/10.3390/ijms19103049
APA StyleWarren, G. M., Stein, R. A., Mchaourab, H. S., & Eichman, B. F. (2018). Movement of the RecG Motor Domain upon DNA Binding Is Required for Efficient Fork Reversal. International Journal of Molecular Sciences, 19(10), 3049. https://doi.org/10.3390/ijms19103049