Designed Elastic Networks: Models of Complex Protein Machinery



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Design of Functional Elastic Networks
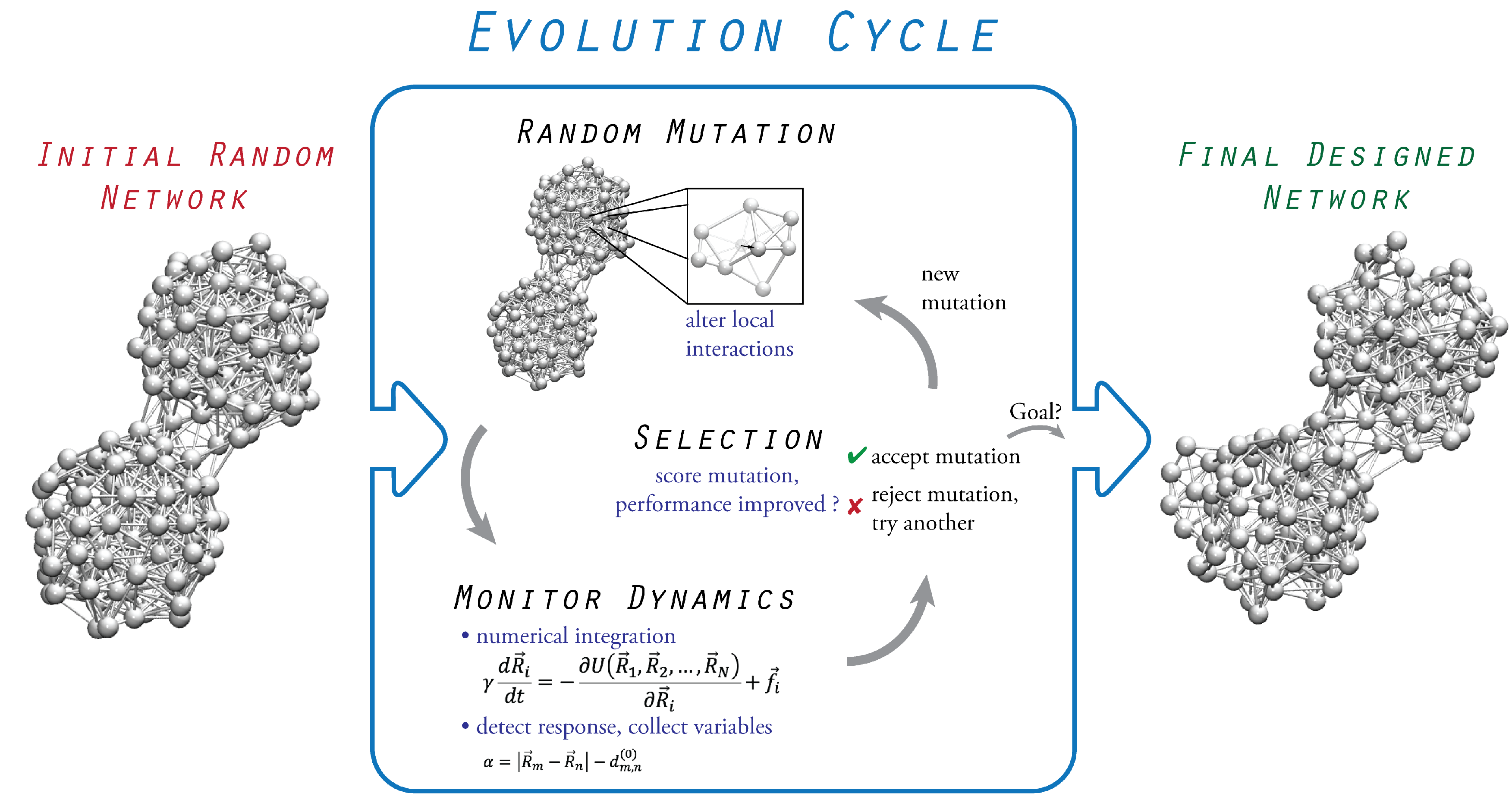
3. Evolutionary Optimization
3.1. Initial Random Network
3.2. Probing of Network Dynamics
3.3. Evolution Cycle
3.4. Termination
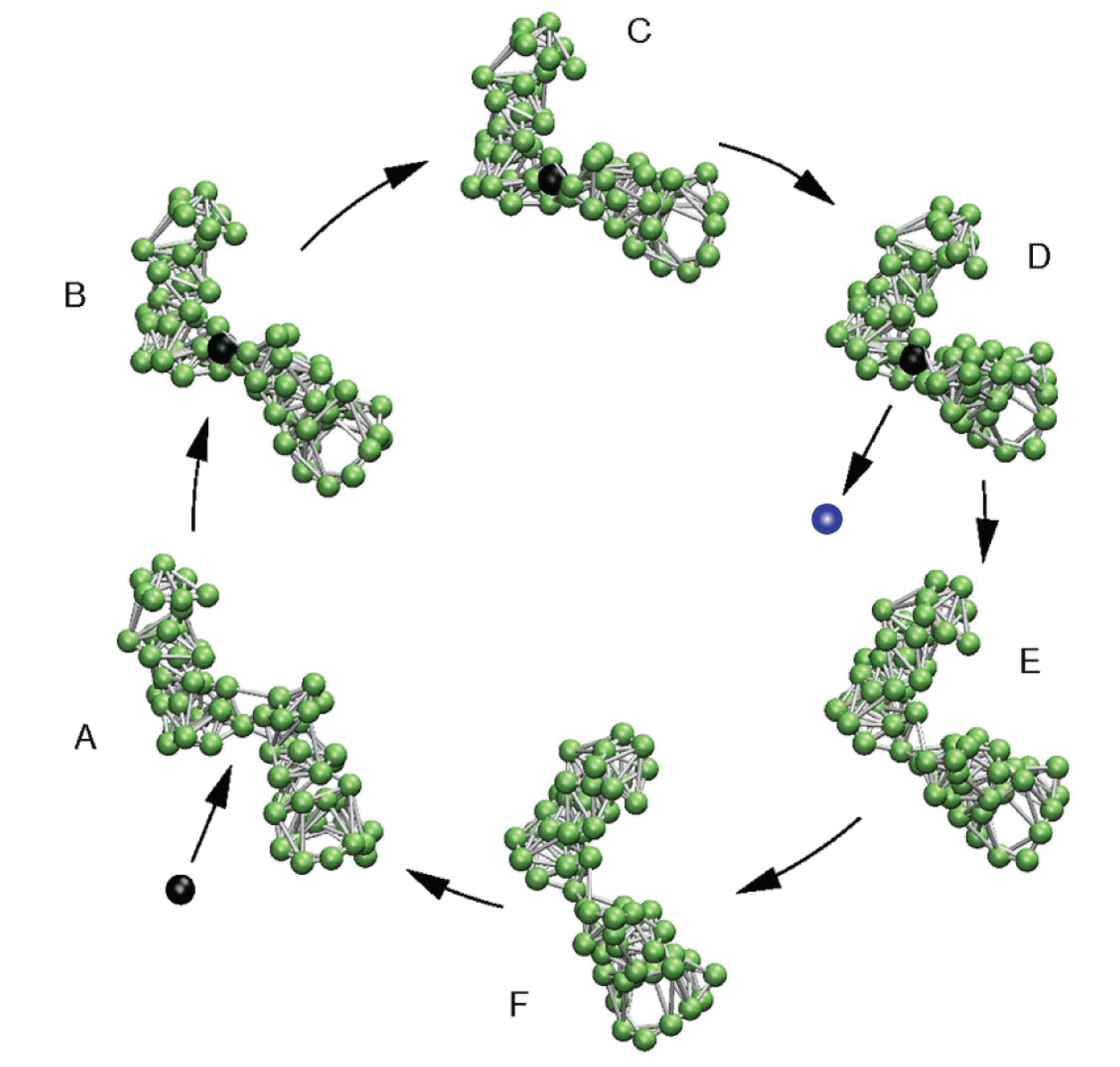
4. Model Molecular Machine
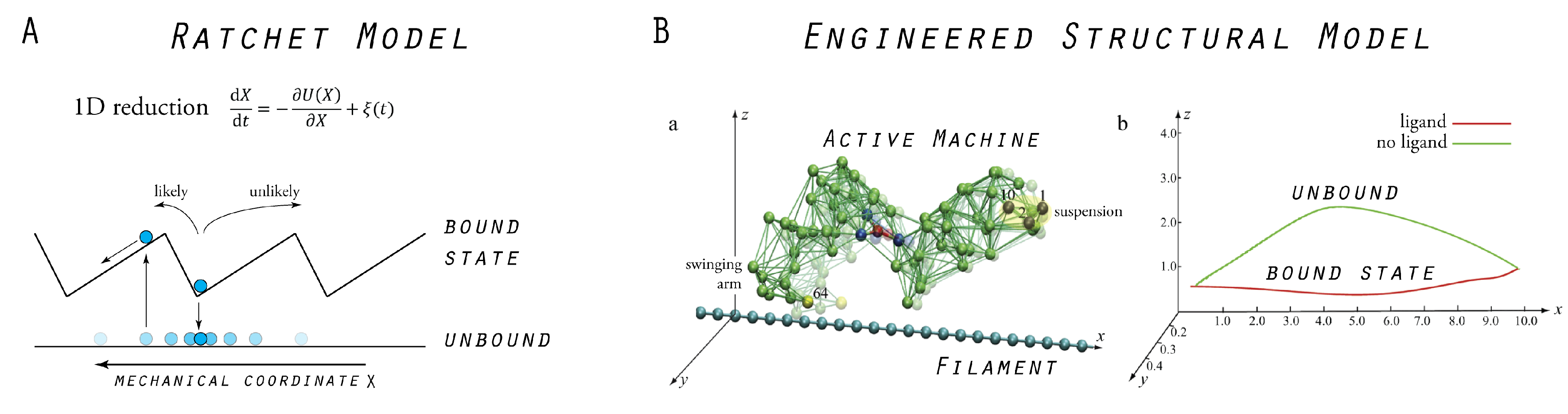
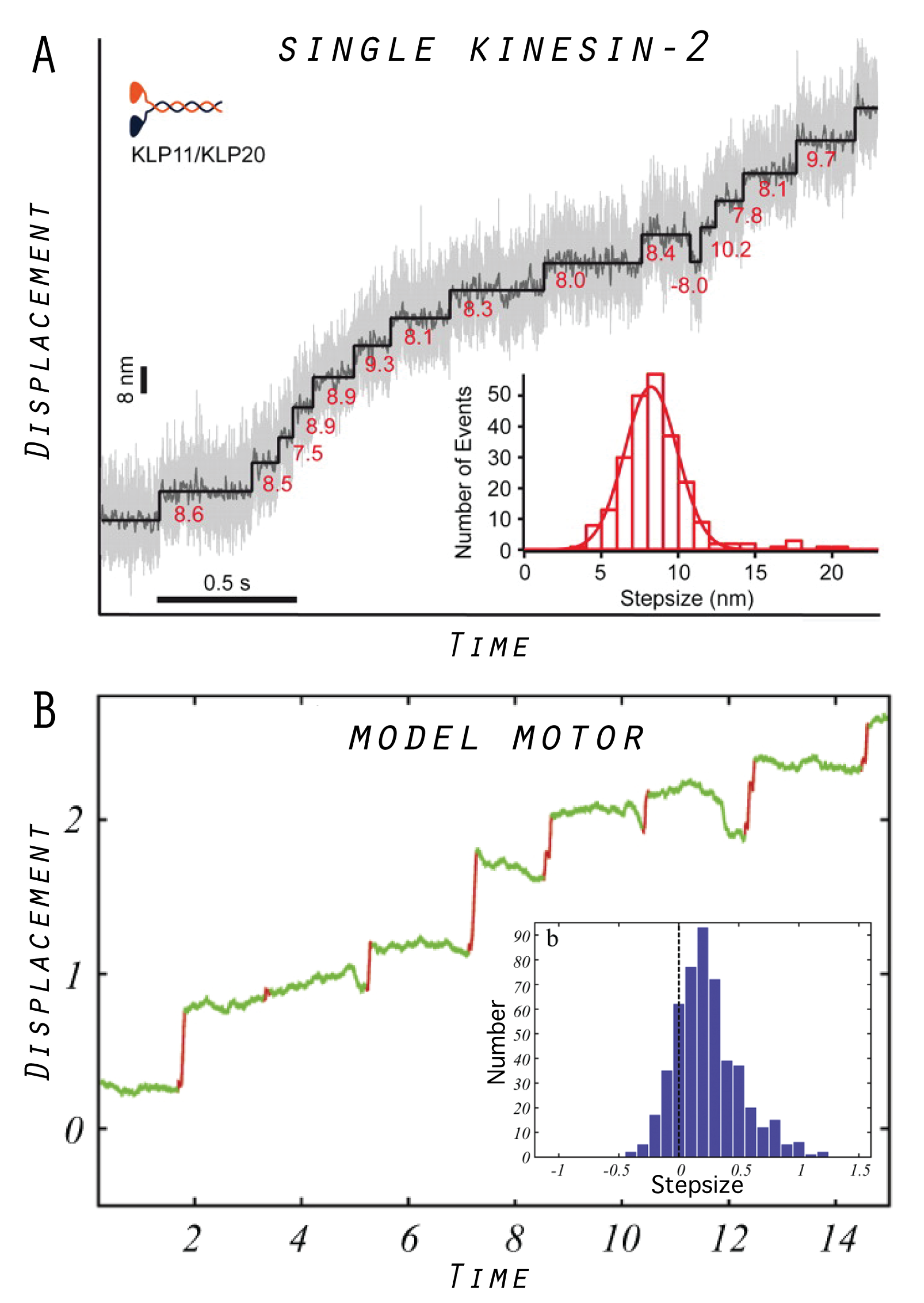
5. Model Molecular Motor
5.1. Tight Coupling Mechanism—Power Stroke Ratcheting Mode
5.2. Loose Coupling Mechanism—Brownian Ratchet Mode
5.3. Designed Motor as a Model System
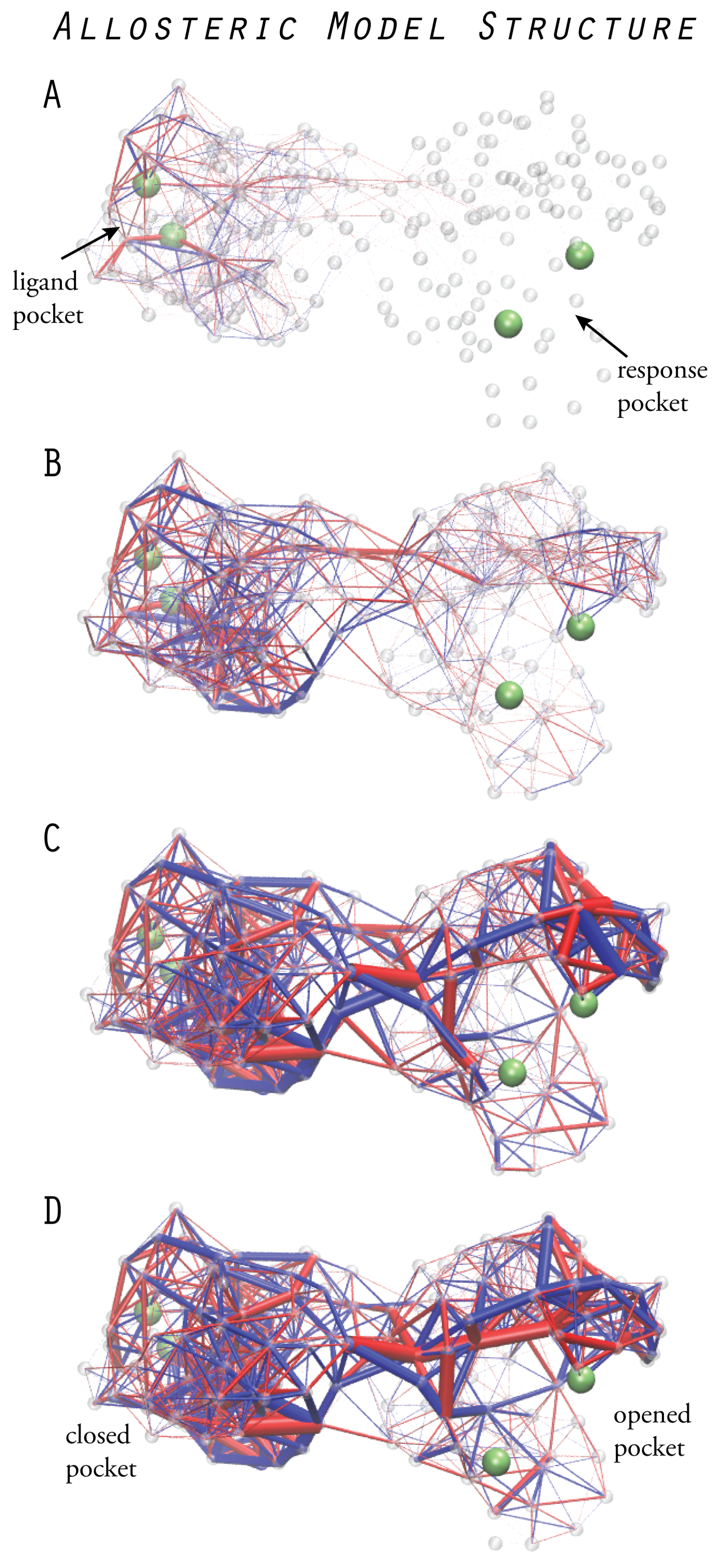
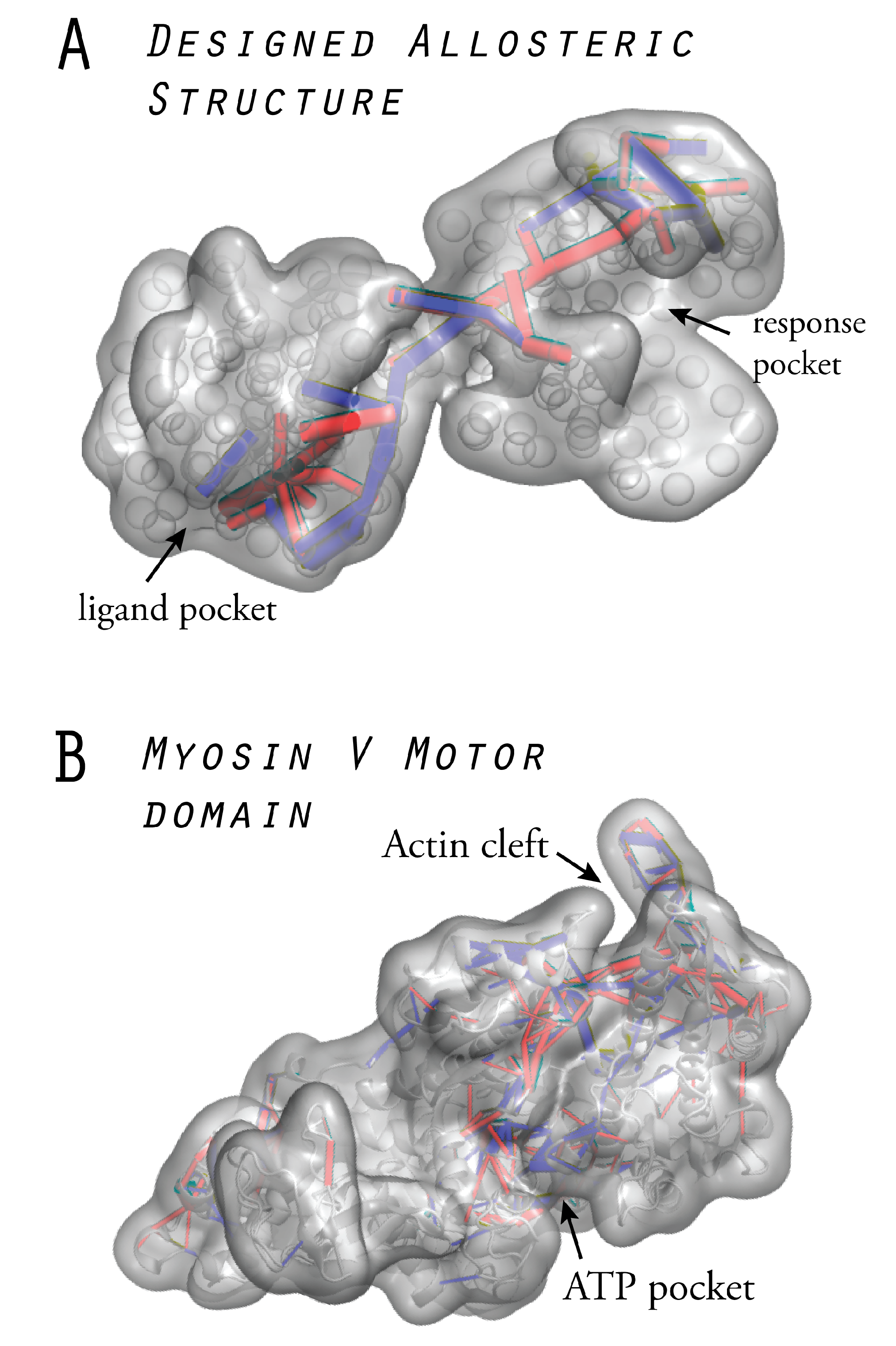
6. Models of Allosteric Protein Machines
6.1. Design of Allosteric Network Structures
6.2. Prototype Allosteric Structures
6.3. Mechanical Networks with Allosteric Interactions
7. Discussion
7.1. Evolution Model: Autonomously Learning Structures and Dimensionality Reduction
7.2. Structurally Resolved Model Systems of Protein Machinery
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 1998, 92, 291–294. [Google Scholar] [CrossRef]
- Mikhailov, A.S.; Ertl, G. Molecular machines. In Chemical Complexity, Self-Organization Processes in Molecular Systems; The Frontiers Collection; Springer: Berlin, Germany, 2017; pp. 181–202. [Google Scholar]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997, 79, 3090–3093. [Google Scholar] [CrossRef]
- Tama, F.; Sanejouand, Y.H. Conformational change of proteins arising from normal mode calculations. Prot. Eng. 2001, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Doniach, S. A comparative study of motor-protein motions by using a simple elastic-network model. Proc. Natl. Acad. Sci. USA 2003, 100, 13253–13258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Song, G.; Jernigan, R.L. How well can we understand large-scale protein motions using normal modes of elastic networks. Biophys. J. 2007, 93, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Mikhailov, A.S. Nonlinear relaxation dynamics in elastic networks and design principles of molecular machines. Proc. Natl. Acad. Sci. USA 2007, 104, 8697–8702. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, H.; Mikhailov, A.S. Tracing entire operation cycles of molecular motor hepatitis C virus helicase in structurally resolved dynamical simulations. Proc. Natl. Acad. Sci. USA 2010, 107, 20875–20880. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Yanagida, T.; Mikhailov, A.S. Nonlinearity of mechanochemical motions in motor proteins. PLoS Comput. Biol. 2010, 6, e1000814. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, H.; Popp, D.; Mikhailov, A.S. In silico investigation of conformational motions in superfamily 2 helicase proteins. PLoS ONE 2011, 6, e21809. [Google Scholar] [CrossRef] [PubMed]
- Düttmann, M.; Togashi, Y.; Yanagida, T.; Mikhailov, A.S. Myosin-V as a mechanical sensor: An elastic network study. Biophys. J. 2012, 102, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, H. Computational biology approach to uncover hepatitis C virus helicase operation. World J. Gastroenterol. 2014, 20, 3401–3409. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, H. TALEs from a spring–superelasticity of Tal effector protein structures. PLoS ONE 2014, 9, e109919. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, H. Nucleotide-induced conformational dynamics in ABC transporters from structure-based coarse grained modeling. Front. Phys. 2016, 4, 3. [Google Scholar] [CrossRef]
- Dai, L.; Flechsig, H.; Yu, J. Deciphering intrinsic inter-subunit couplings that lead to sequential hydrolysis of F1-ATPase ring. Biophys. J. 2017, 113, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Rocks, J.W.; Pashine, N.; Bischofberger, I.; Goodrich, C.P.; Liu, A.J.; Nagel, S.R. Designing allostery-inspired response in mechanical networks. Proc. Natl. Acad. Sci. USA 2017, 114, 2520–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Ravasio, R.; Brito, C.; Wyart, M. Architecture and coevolution of allosteric materials. Proc. Natl. Acad. Sci. USA 2017, 114, 2526–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flechsig, H. Design of elastic networks with evolutionary optimised long-range communication as mechanical models of allosteric proteins. Biophys. J. 2017, 113, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Eckmann, J.-P.; Libchaber, A.; Tlusty, T. Green function of correlated genes in a minimal mechanical model of protein evolution. Proc. Natl. Acad. Sci. USA 2018, 115, E4559–E4568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Flechsig, H.; Mikhailov, A.S. Towards synthetic molecular motors: A model elastic-network study. New J. Phys. 2016, 18, 043006. [Google Scholar] [CrossRef]
- Atilgan, C.; Gerek, Z.N.; Ozkan, S.B.; Atilgan, A.R. Manipulation of conformational change in proteins by single-residue perturbations. Biophys. J. 2010, 99, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Cressman, A.; Togashi, Y.; Mikhailov, A.S.; Kapral, R. Mesoscale modeling of molecular machines: Cyclic dynamics and hydrodynamical fluctuations. Phys. Rev. E 2008, 77, 050901(R). [Google Scholar] [CrossRef] [PubMed]
- Huang, M.J.; Kapral, R.; Mikhailov, A.S.; Chen, H.-Y. Coarse-grain simulations of active molecular machines in lipid bilayers. J. Chem. Phys. 2013, 138, 195101. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. How molecular motors work. Nature 1994, 372, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D.; Milligan, R.A. The way things move: Looking under the hood of molecular motor proteins. Science 2000, 288, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Cordova, N.J.; Ermentrout, B.; Oster, G. Dynamics of single-motor molecules: The thermal ratchet model. Proc. Natl. Acad. Sci. USA 1992, 89, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Astumian, R.D.; Bier, M. Fluctuation driven ratchets: Molecular motors. Phys. Rev. Lett. 1994, 72, 1766–1769. [Google Scholar] [CrossRef] [PubMed]
- Jülicher, F.; Ajdari, A.; Prost, J. Modeling molecular motors. Rev. Mod. Phys. 1997, 69, 917–922. [Google Scholar] [CrossRef]
- Brunnbauer, M.; Mueller-Planitz, F.; Kösem, S.; Hieu Ho, T.; Dombi, R.; Gebhardt, J.C.M.; Rief, M.; Ökten, Z. Regulation of a heterodimeric kinesin-2 through an unprocessive motor domain that is turned processive by its partner. Proc. Natl. Acad. Sci. USA 2010, 107, 10460–10465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, A.; Berendzen, J.; Bowne, S.F.; Frauenfelder, H.; Iben, I.E.; Sauke, T.B.; Shyamsunder, E.; Young, R.D. Protein states and proteinquakes. Proc. Natl. Acad. Sci. USA 1985, 85, 5000–5004. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Nussinov, R. A unified view of how allostery works. PLoS Comput. Biol. 2014, 10, e1003394. [Google Scholar] [CrossRef] [PubMed]
- Brüschweiler, S.; Schanda, P.; Kloiber, K.; Brutscher, B.; Kontaxis, G.; Konrat, R.; Trollinger, M. Direct observation of the dynamic process underlying allosteric signal transmission. J. Am. Chem. Soc. 2009, 131, 3063–3068. [Google Scholar] [CrossRef] [PubMed]
- Grutsch, S.; Brüschweiler, S.; Trollinger, M. NMR methods to study dynamic allostery. PLoS Comput. Biol. 2016, 12, e1004620. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Tobi, D.; Bahar, I. Allosteric changes in protein structure computed by a simple mechanical model: Hemoglobin T-R2 transition. J. Mol. Biol. 2003, 333, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Chennubhotla, C.; Tobi, D. Intrinsic dynamics of enzymes in the unbound state and relation to allosteric regulation. Curr. Opin. Struct. Biol. 2007, 17, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Majek, P.; Bahar, I. Allosteric transitions of supramolecular systems explored by network models: Application to chaperonin GroEL. PLoS Comput. Biol. 2009, 5, e1000360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erman, B. A fast approximate method of identifying paths of allosteric communication in proteins. Proteins 2013, 81, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.-Q.; Skjaerven, L.; Grant, B.J. Rapid characterization of allosteric networks with ensemble normal mode analysis. J. Phys. Chem. B 2016, 120, 8276–8288. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, A.; Erman, B. Causality, transfer entropy, and allosteric communication landscapes in proteins with harmonic interactions. Proteins 2017, 85, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Hexner, D.; Liu, A.J.; Nagel, S.R. Role of local response in manipulating the elastic properties of disordered solids by bond removal. Soft Matter 2018, 14, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocks, J.W.; Ronellenfitsch, H.; Liu, A.J.; Nagel, S.R.; Katifori, E. The limits of multifunctionality in tunable networks. arXiv, 2018; arXiv:1805.00504v1. [Google Scholar]
- Borovinskiy, A.L.; Grosberg, A.Y. Design of toy proteins capable of rearranging conformations in a mechanical fashion. J. Chem. Phys. 2003, 118, 5201. [Google Scholar] [CrossRef] [Green Version]
- Tlusty, T.; Libchaber, A.; Eckmann, J.-P. Physical model of the genotype-to-phenotype map of proteins. Phys. Rev. X 2017, 7, 021037. [Google Scholar] [CrossRef]
- Yan, L.; Ravasio, R.; Brito, C.; Wyart, M. Principles for optimal cooperativity in allosteric materials. Biophys. J. 2018, 114, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Webb, M.R.; Forgacs, E.; White, H.D.; Sellers, J.R. Direct observation of the mechanochemical coupling in myosin Va during processive movement. Nature 2008, 455, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, M.; Takagi, H.; Shibata, T.; Iwane, A.H.; Yanagida, T. Fluctuation analysis of mechanochemical coupling depending on the type of biomolecular motors. Phys. Rev. Lett. 2008, 101, 128103. [Google Scholar] [CrossRef] [PubMed]
- Oosawa, F. The loose coupling mechanism in molecular machines of living cells. Genes Cells 2000, 5, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagida, T.; Arata, T.; Oosawa, F. Sliding distance of actin filaments induced by a myosin cross-bridge during one ATP hydrolysis cycle. Nature 1985, 316, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Koshland, D.E.; Nemethy, G.; Filmer, D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 1966, 5, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Tatsumi-Koga, R.; Liu, G.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Baker, D. Principles for designing ideal protein structures. Nature 2012, 491, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flechsig, H.; Togashi, Y. Designed Elastic Networks: Models of Complex Protein Machinery. Int. J. Mol. Sci. 2018, 19, 3152. https://doi.org/10.3390/ijms19103152
Flechsig H, Togashi Y. Designed Elastic Networks: Models of Complex Protein Machinery. International Journal of Molecular Sciences. 2018; 19(10):3152. https://doi.org/10.3390/ijms19103152
Chicago/Turabian StyleFlechsig, Holger, and Yuichi Togashi. 2018. "Designed Elastic Networks: Models of Complex Protein Machinery" International Journal of Molecular Sciences 19, no. 10: 3152. https://doi.org/10.3390/ijms19103152
APA StyleFlechsig, H., & Togashi, Y. (2018). Designed Elastic Networks: Models of Complex Protein Machinery. International Journal of Molecular Sciences, 19(10), 3152. https://doi.org/10.3390/ijms19103152