mTOR Complexes as a Nutrient Sensor for Driving Cancer Progression

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
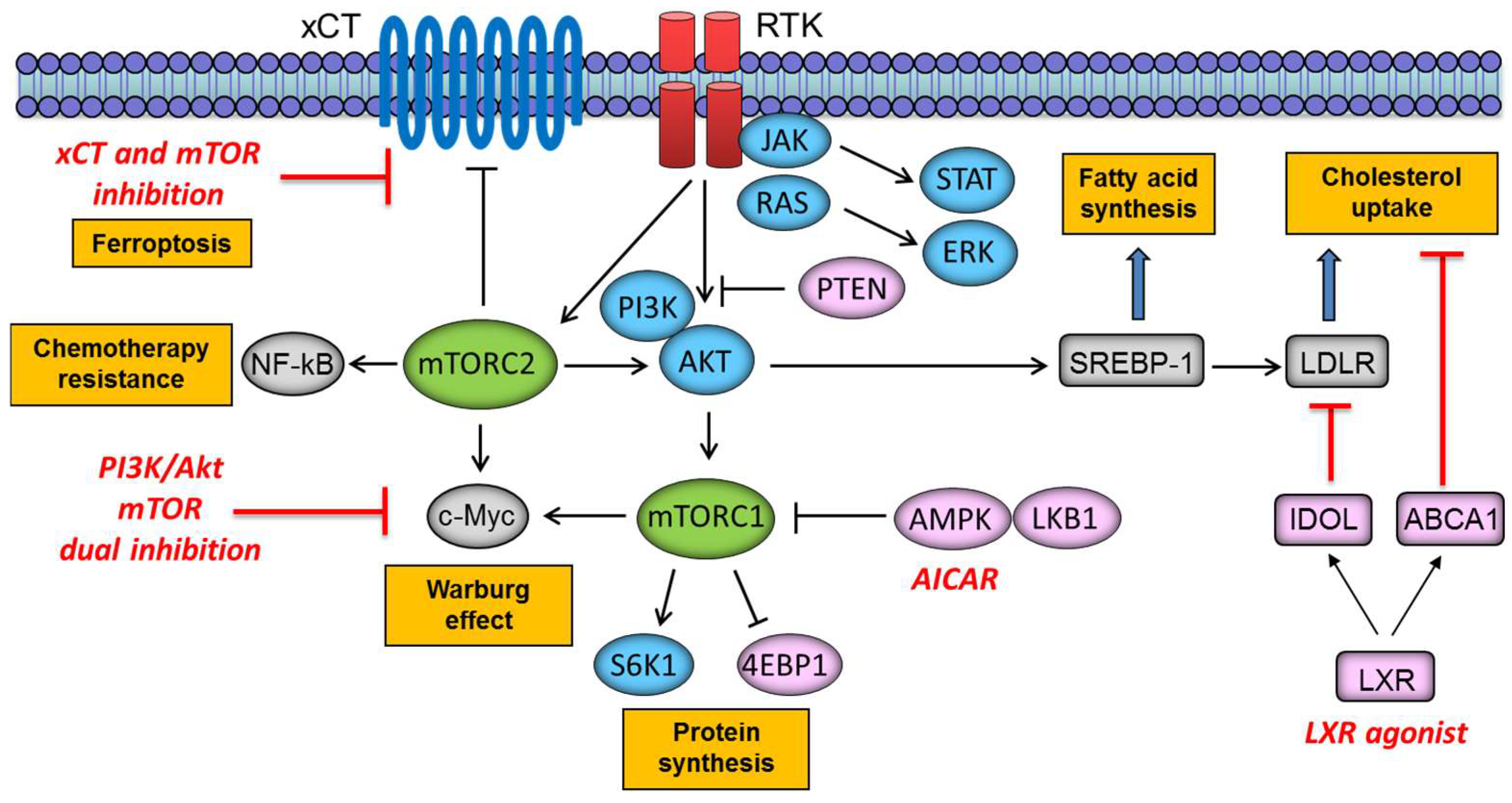{kind=link}
1. Introduction
2. Metabolic Reprogramming as an Essential Hallmark in Cancer
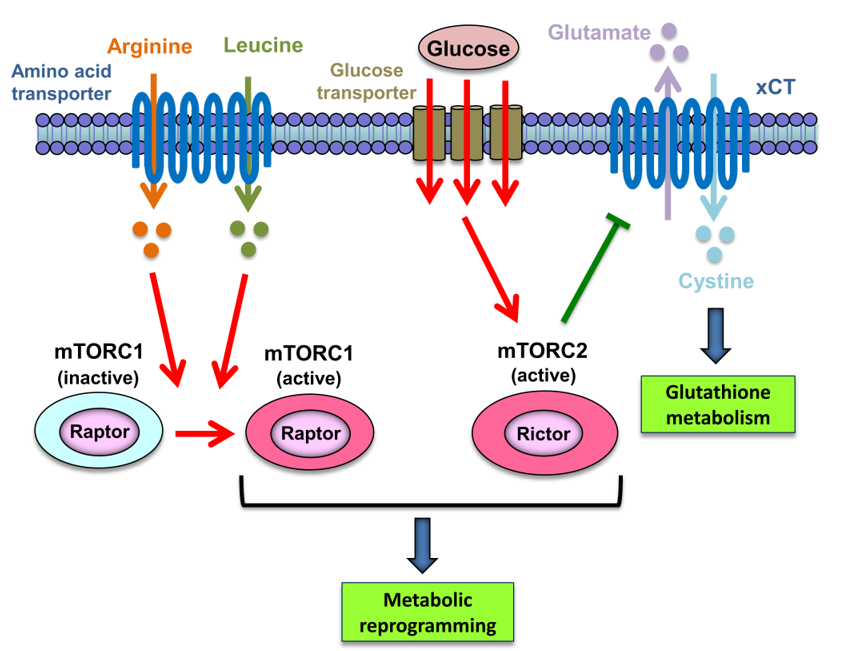
3. mTORC1 and mTORC2-Irreplaceable Partners in Cancer Metabolic Reprogramming
4. mTORC1 as a Sensor of Amino Acids in Cancer Cells
5. mTORC2 at the Intersection of Glucose and Amino Acid Metabolism
6. Epigenetic Modulation by mTOR-Dependent Metabolism in Cancer
7. Molecular Therapies Targeting mTOR-Dependent Signaling and Metabolism
8. Unanswered Questions on mTOR-Dependent Metabolism in Cancer
9. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Soga, T. Cancer metabolism: Key players in metabolic reprogramming. Cancer Sci. 2013, 104, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Energy boost: The warburg effect returns in a new theory of cancer. J. Natl. Cancer Inst. USA 2004, 96, 1805–1806. [Google Scholar] [CrossRef] [PubMed]
- Miyo, M.; Konno, M.; Nishida, N.; Sueda, T.; Noguchi, K.; Matsui, H.; Colvin, H.; Kawamoto, K.; Koseki, J.; Haraguchi, N.; et al. Metabolic adaptation to nutritional stress in human colorectal cancer. Sci. Rep. 2016, 6, 38415. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Ueda-Wakagi, M.; Sato, T.; Kawasaki, K.; Sawada, K.; Kawabata, K.; Akagawa, M.; Ashida, H. Measurement of glucose uptake in cultured cells. Curr. Protoc. Pharmacol. 2015, 71, 12.14.1–12.14.26. [Google Scholar] [PubMed]
- Rock, C.L.; Doyle, C.; Demark-Wahnefried, W.; Meyerhardt, J.; Courneya, K.S.; Schwartz, A.L.; Bandera, E.V.; Hamilton, K.K.; Grant, B.; McCullough, M.; et al. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J. Clin. 2012, 62, 243–274. [Google Scholar] [CrossRef] [PubMed]
- Izuishi, K.; Kato, K.; Ogura, T.; Kinoshita, T.; Esumi, H. Remarkable tolerance of tumor cells to nutrient deprivation: Possible new biochemical target for cancer therapy. Cancer Res. 2000, 60, 6201–6207. [Google Scholar] [PubMed]
- Davis, C.D. Nutritional interactions: Credentialing of molecular targets for cancer prevention. Exp. Biol. Med. (Maywood) 2007, 232, 176–183. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. Ros homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Chen, M.; Cheng, X.; Qi, Y.; Chen, Y.; Das, I.; Li, X.; Vallat, B.; Fu, L.W.; Qian, C.N.; et al. SOD1 phosphorylation by mTOR1 couples nutrient sensing and redox Regulation. Mol. Cell 2018, 70, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab. 2014, 25, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic reprogramming by the PI3K-AKT-mTOR pathway in cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar] [PubMed]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and nutrient homeostasis: The central role of the lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Chou, C.C.; Chuang, H.C.; Hsu, E.C.; Chiu, P.C.; Kulp, S.K.; Byrd, J.C.; Chen, C.S. Functional role of mTORC2 versus integrin-linked kinase in mediating ser473-akt phosphorylation in pten-negative prostate and breast cancer cell lines. PLoS ONE 2013, 8, e67149. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Piao, H.L.; Zhuang, L.; Sarbassov dos, D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Im-aram, A.; Farrand, L.; Bae, S.M.; Song, G.; Song, Y.S.; Han, J.Y.; Tsang, B.K. The mTORC2 component rictor contributes to cisplatin resistance in human ovarian cancer cells. PLoS ONE 2013, 8, e75455. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; De Jesus, J.; et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Blaes, J.; Weiler, M. mTORC 2:1 for chemotherapy sensitization in glioblastoma. Cancer Discov. 2011, 1, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian tor complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Buel, G.R.; Blenis, J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol. Cells 2013, 35, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. Deptor is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates tor action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jeno, P.; Arrieumerlou, C.; Hall, M.N. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE 2007, 2, e1217. [Google Scholar] [CrossRef] [PubMed]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the AKT/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. Msin1 is necessary for AKT/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates AKT phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of protor as a novel rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Sommer, E.M.; Sakamoto, K.; Wullschleger, S.; Alessi, D.R. Protor-1 is required for efficient mTORC2-mediated activation of sgk1 in the kidney. Biochem. J. 2011, 436, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Leon-Annicchiarico, C.L.; Munoz-Pinedo, C. Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol. 2014, 542, 59–80. [Google Scholar] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Samanta, A.; Cheng, H.S.; Ding, Z.; Han, W.; Toschi, L.; Chang, Y.T. Effect of oncogene activating mutations and kinase inhibitors on amino acid metabolism of human isogenic breast cancer cells. Mol. Biosyst. 2015, 11, 3378–3386. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; Zhi, J.; Moll, U.M.; Nemajerova, A.; Petrenko, O. Direct reprogramming by oncogenic ras and myc. Proc. Natl. Acad. Sci. USA 2013, 110, 3937–3942. [Google Scholar] [CrossRef] [PubMed]
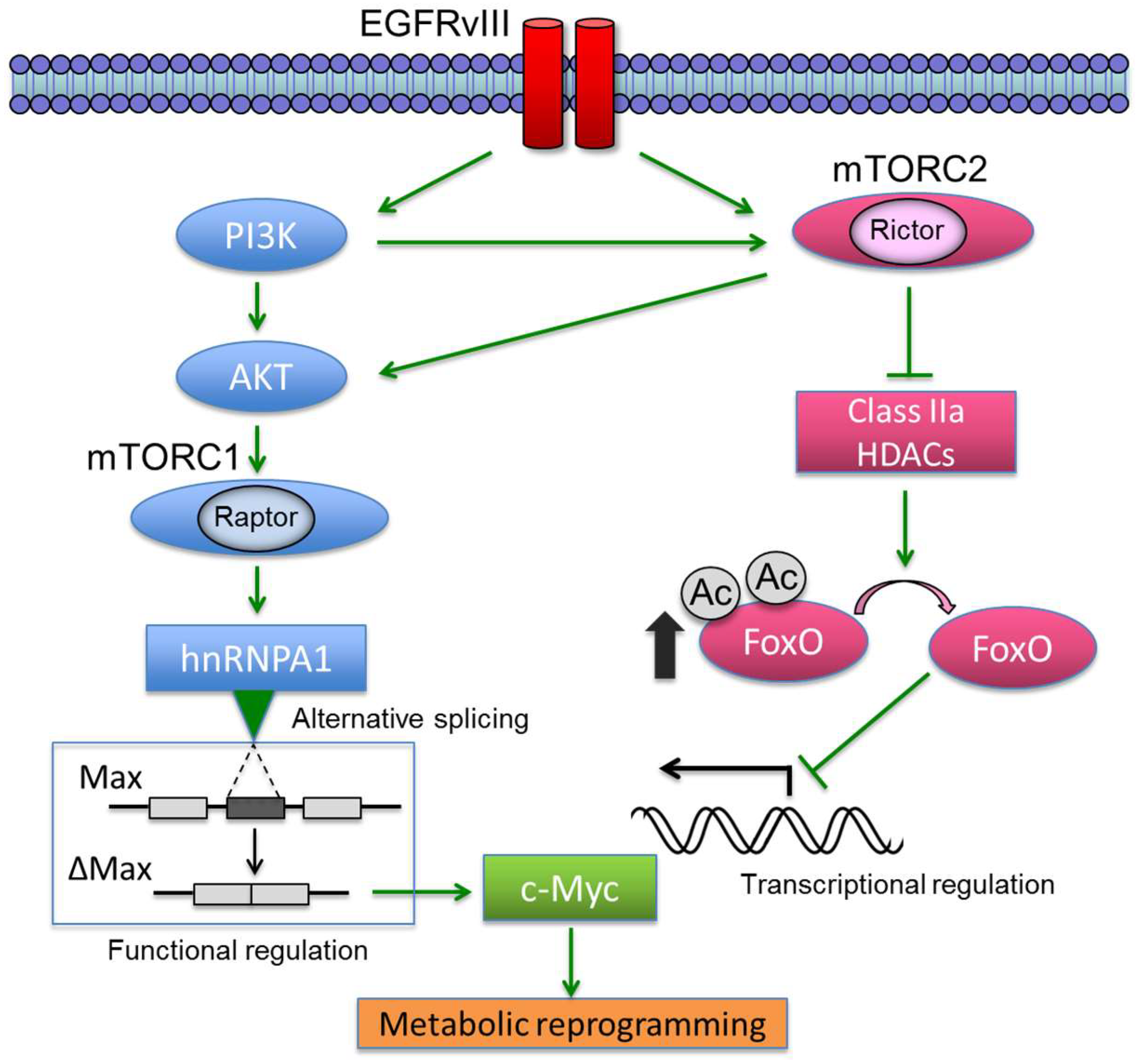
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through foxo acetylation and upregulation of c-myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, C.A. The role of mTORC1 in regulating protein synthesis and skeletal muscle mass in response to various mechanical stimuli. Rev. Physiol. Biochem. Pharmacol. 2014, 166, 43–95. [Google Scholar] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. mTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Blommaart, E.F.; Luiken, J.J.; Blommaart, P.J.; van Woerkom, G.M.; Meijer, A.J. Phosphorylation of ribosomal protein s6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995, 270, 2320–2326. [Google Scholar] [CrossRef] [PubMed]
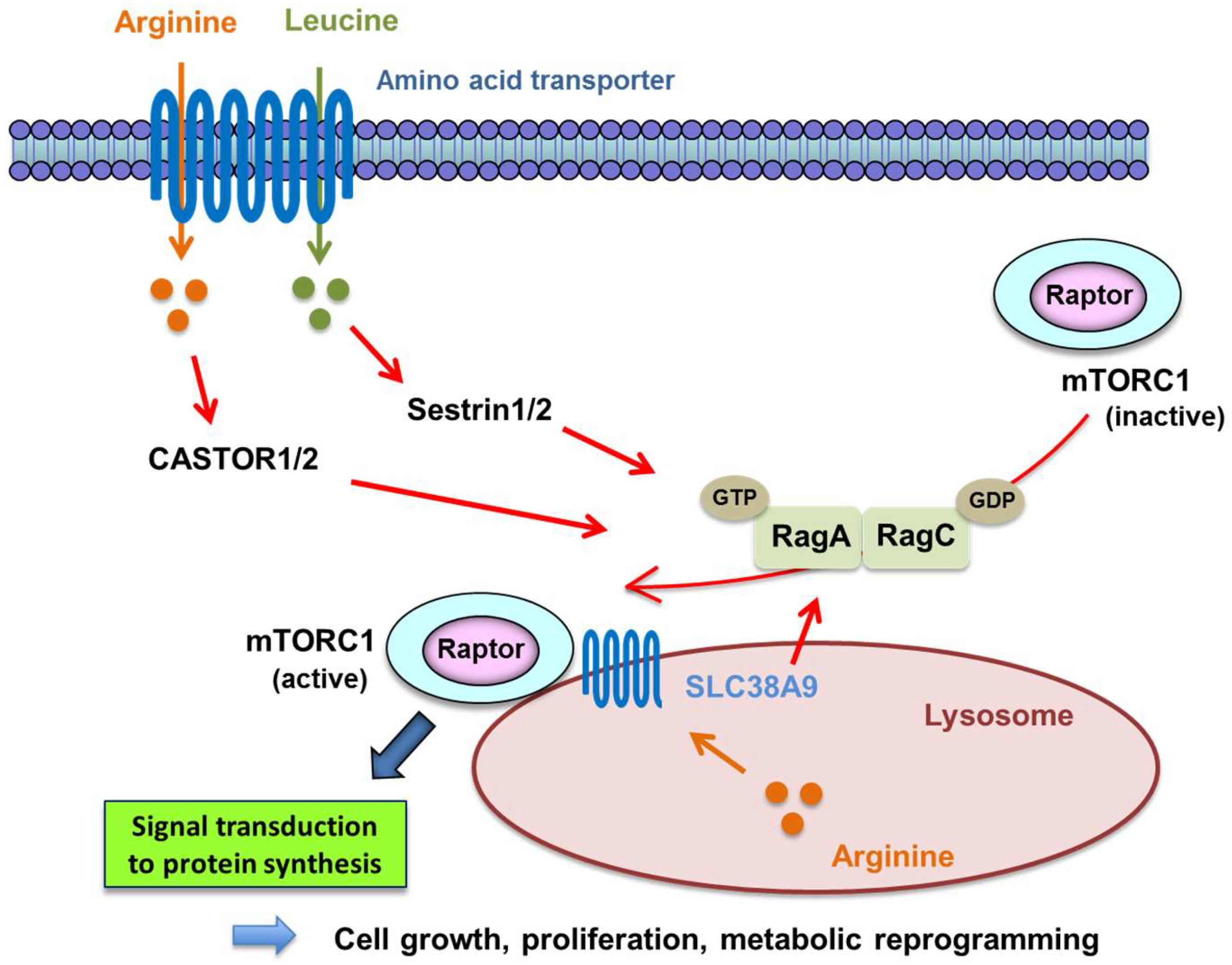
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Potier, M.; Darcel, N.; Tome, D. Protein, amino acids and the control of food intake. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Panten, U.; Christians, J.; von Kriegstein, E.; Poser, W.; Hasselblatt, A. Studies on the mechanism of l-leucine-and alpha-ketoisocaproic acid-induced insulin release from perifused isolated pancreatic islets. Diabetologia 1974, 10, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Greiwe, J.S.; Kwon, G.; McDaniel, M.L.; Semenkovich, C.F. Leucine and insulin activate p70 s6 kinase through different pathways in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E466–E471. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.S.; Schwartz, R.G.; Welle, S. Leucine as a regulator of whole body and skeletal muscle protein metabolism in humans. Am. J. Physiol. 1992, 263, E928–E934. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, J.H.; Yoon, I.; Lee, C.; Fallahi Sichani, M.; Kang, J.S.; Kang, J.; Guo, M.; Lee, K.Y.; Han, G.; et al. Coordination of the leucine-sensing Rag GTPase cycle by leucyl-tRNA synthetase in the mTORC1 signaling pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E5279–E5288. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The castor proteins are arginine sensors for the mTORC1 pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ou, Y.; Yang, Y.; Li, W.; Xu, Y.; Xie, Y.; Liu, Y. Klhl22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature 2018, 557, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORc1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.F. Amino acid sensing mechanisms: An achilles heel in cancer? FEBS J. 2012, 279, 2624–2631. [Google Scholar] [CrossRef] [PubMed]
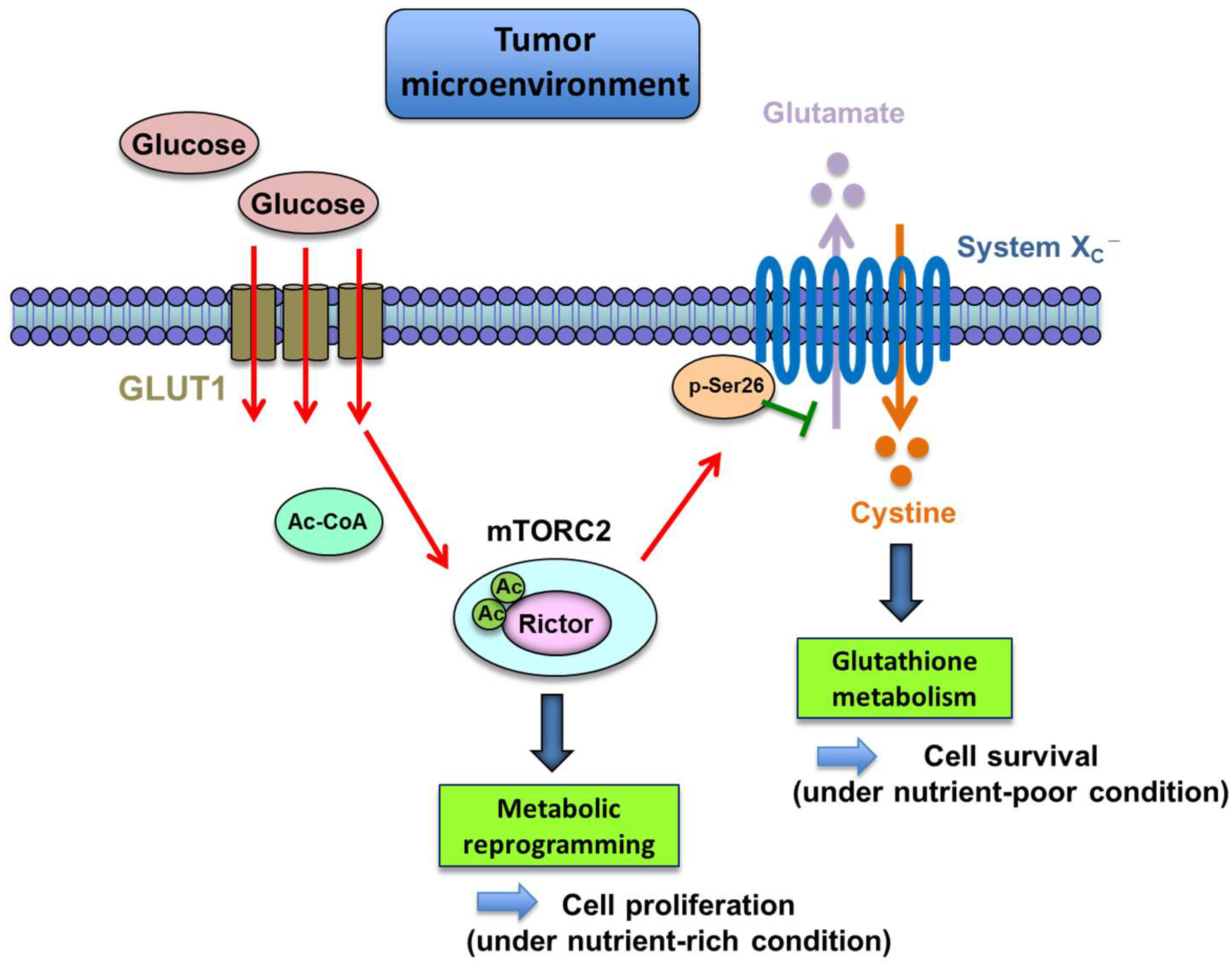
- Masui, K.; Tanaka, K.; Ikegami, S.; Villa, G.R.; Yang, H.; Yong, W.H.; Cloughesy, T.F.; Yamagata, K.; Arai, N.; Cavenee, W.K.; et al. Glucose-dependent acetylation of rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Korkes, S.; Del Campillo, A.; Ochoa, S. Pyruvate oxidation system of heart muscle. J. Biol. Chem. 1952, 195, 541–547. [Google Scholar] [PubMed]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter XCT. Mol. Cell 2017, 67, 128–138.e7. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. mTORC2 and metabolic reprogramming in gbm: At the interface of genetics and environment. Brain Pathol. 2015, 25, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. Atp citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. Atp-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class iia histone deacetylases are hormone-activated regulators of foxo and mammalian glucose homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1c promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Wagner, M.; Reil, J.C.; Müller, S.A.; Tauchnitz, M.; Zimmer, A.M.; Lehmann, L.H.; Thiel, G.; Böhm, M.; Backs, J.; et al. Hdac4 controls histone methylation in response to elevated cardiac load. J. Clin. Investig. 2013, 123, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. Cancer metabolism as a central driving force of glioma pathogenesis. Brain Tumor Pathol. 2016, 33, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual pi3 Kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. mTORC2 dictates warburg effect and drug resistance. Cell Cycle 2014, 13, 1053–1054. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.; Watson, A.D.; Phelps, M.; et al. The ampk agonist aicar inhibits the growth of egfrviii-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an AKT-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An lxr-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.; Salloum, D.; Bernfeld, E.; Gorodetsky, E.; Akselrod, A.; Frias, M.A.; Sudderth, J.; Chen, P.H.; DeBerardinis, R.; Foster, D.A. Lipid sensing by mTOR complexes via de novo synthesis of phosphatidic acid. J. Biol. Chem. 2017, 292, 6303–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, K.; Shibata, N.; Cavenee, W.K.; Mischel, P.S. mTORC2 activity in brain cancer: Extracellular nutrients are required to maintain oncogenic signaling. Bioessays 2016, 38, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harachi, M.; Masui, K.; Okamura, Y.; Tsukui, R.; Mischel, P.S.; Shibata, N. mTOR Complexes as a Nutrient Sensor for Driving Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3267. https://doi.org/10.3390/ijms19103267
Harachi M, Masui K, Okamura Y, Tsukui R, Mischel PS, Shibata N. mTOR Complexes as a Nutrient Sensor for Driving Cancer Progression. International Journal of Molecular Sciences. 2018; 19(10):3267. https://doi.org/10.3390/ijms19103267
Chicago/Turabian StyleHarachi, Mio, Kenta Masui, Yukinori Okamura, Ryota Tsukui, Paul S. Mischel, and Noriyuki Shibata. 2018. "mTOR Complexes as a Nutrient Sensor for Driving Cancer Progression" International Journal of Molecular Sciences 19, no. 10: 3267. https://doi.org/10.3390/ijms19103267
APA StyleHarachi, M., Masui, K., Okamura, Y., Tsukui, R., Mischel, P. S., & Shibata, N. (2018). mTOR Complexes as a Nutrient Sensor for Driving Cancer Progression. International Journal of Molecular Sciences, 19(10), 3267. https://doi.org/10.3390/ijms19103267