Type-II tRNAs and Evolution of Translation Systems and the Genetic Code



Abstract
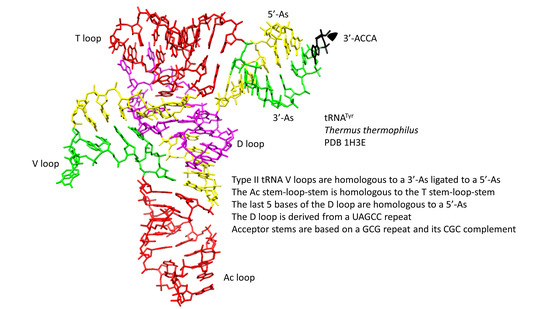
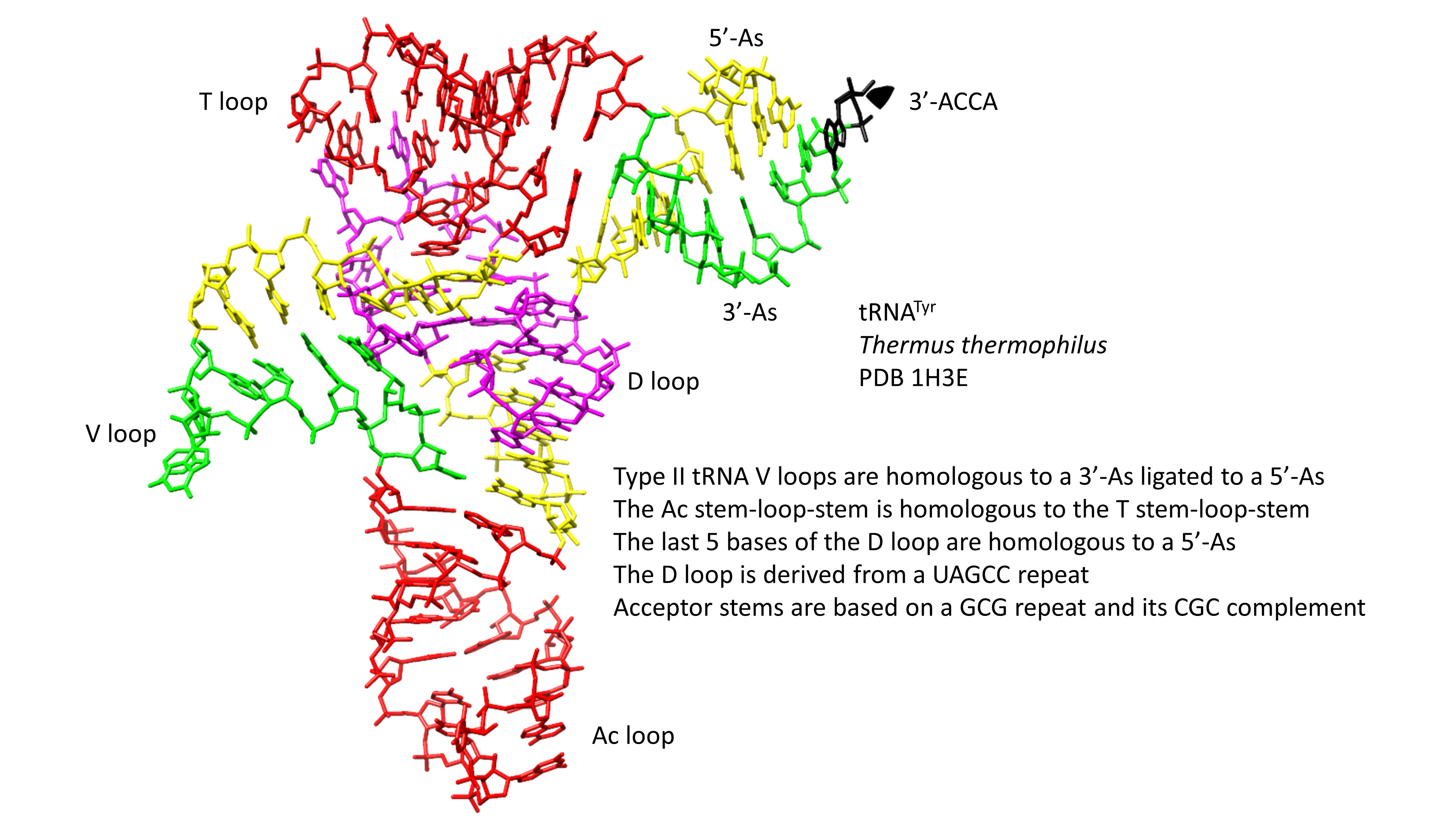
:
1. Introduction
2. Results
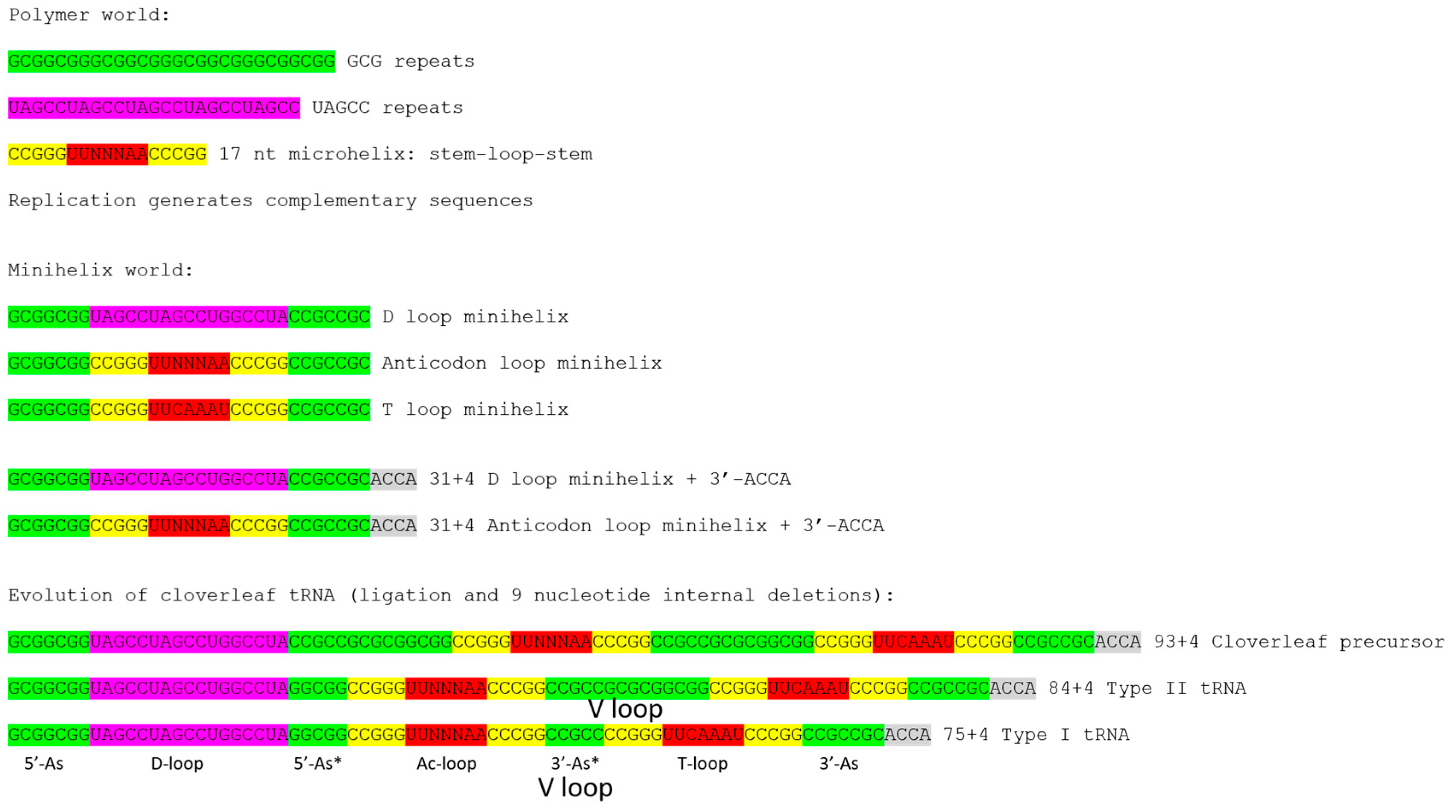
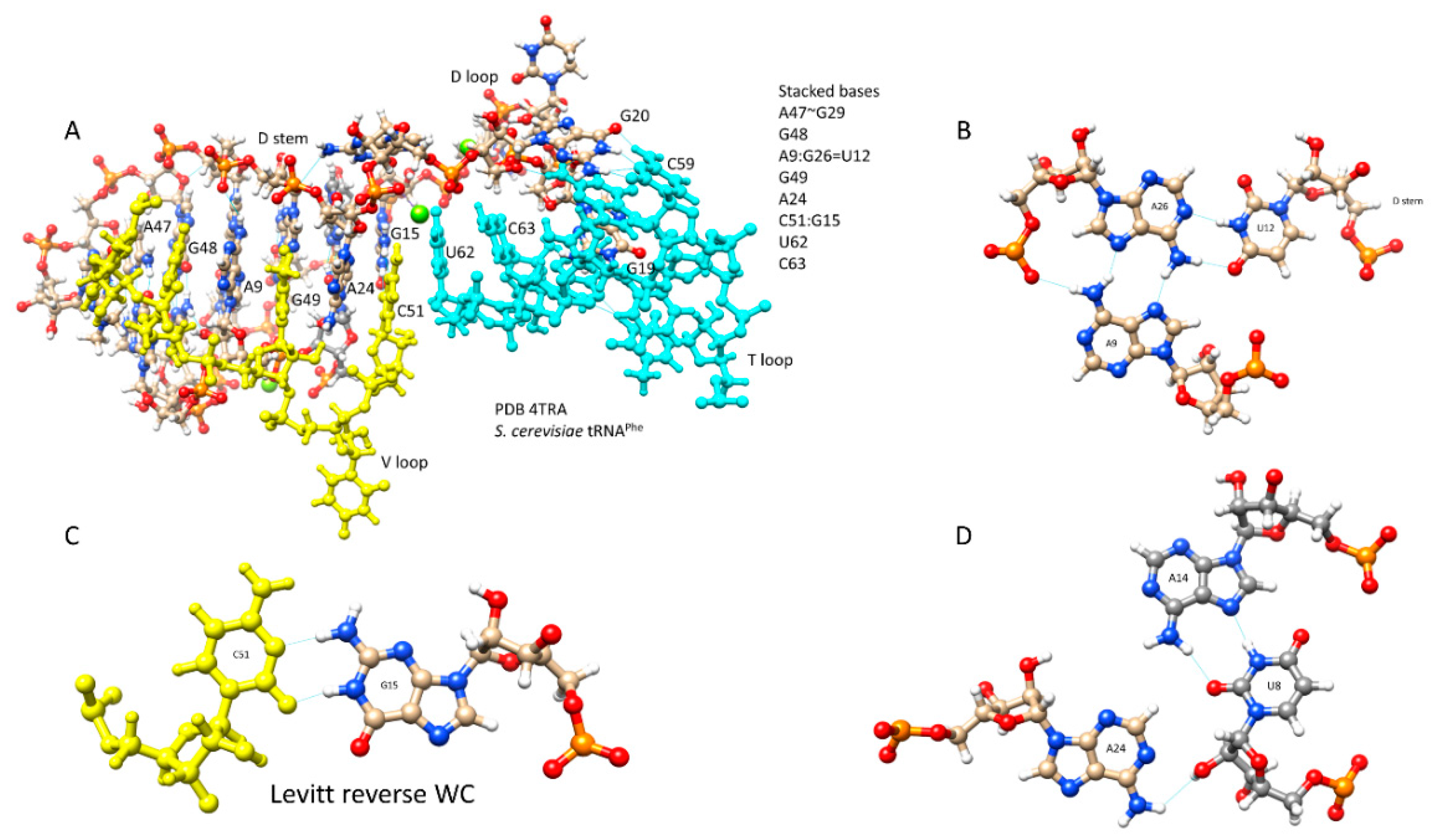
2.1. A Model for Evolution of Type-II tRNAs
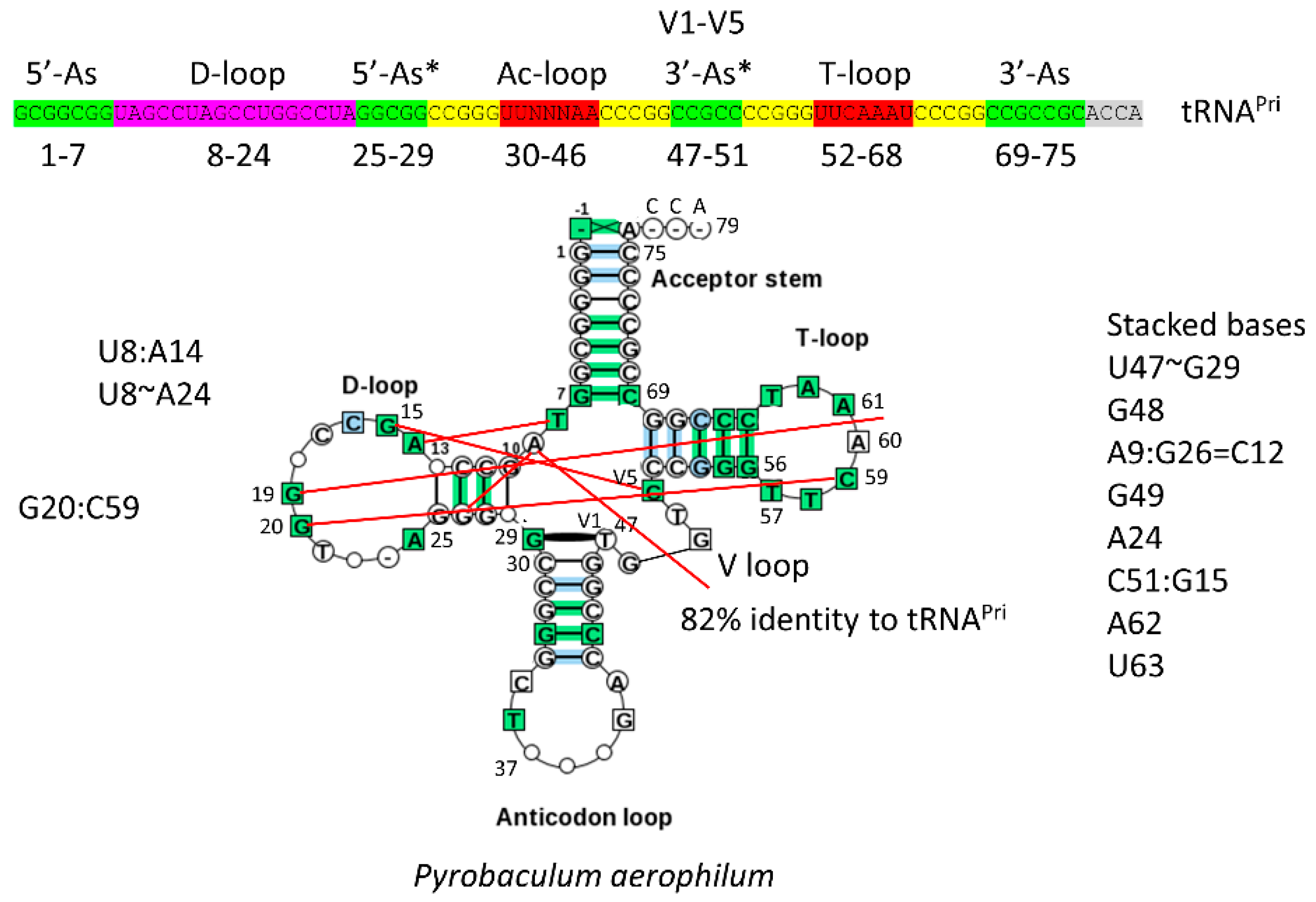
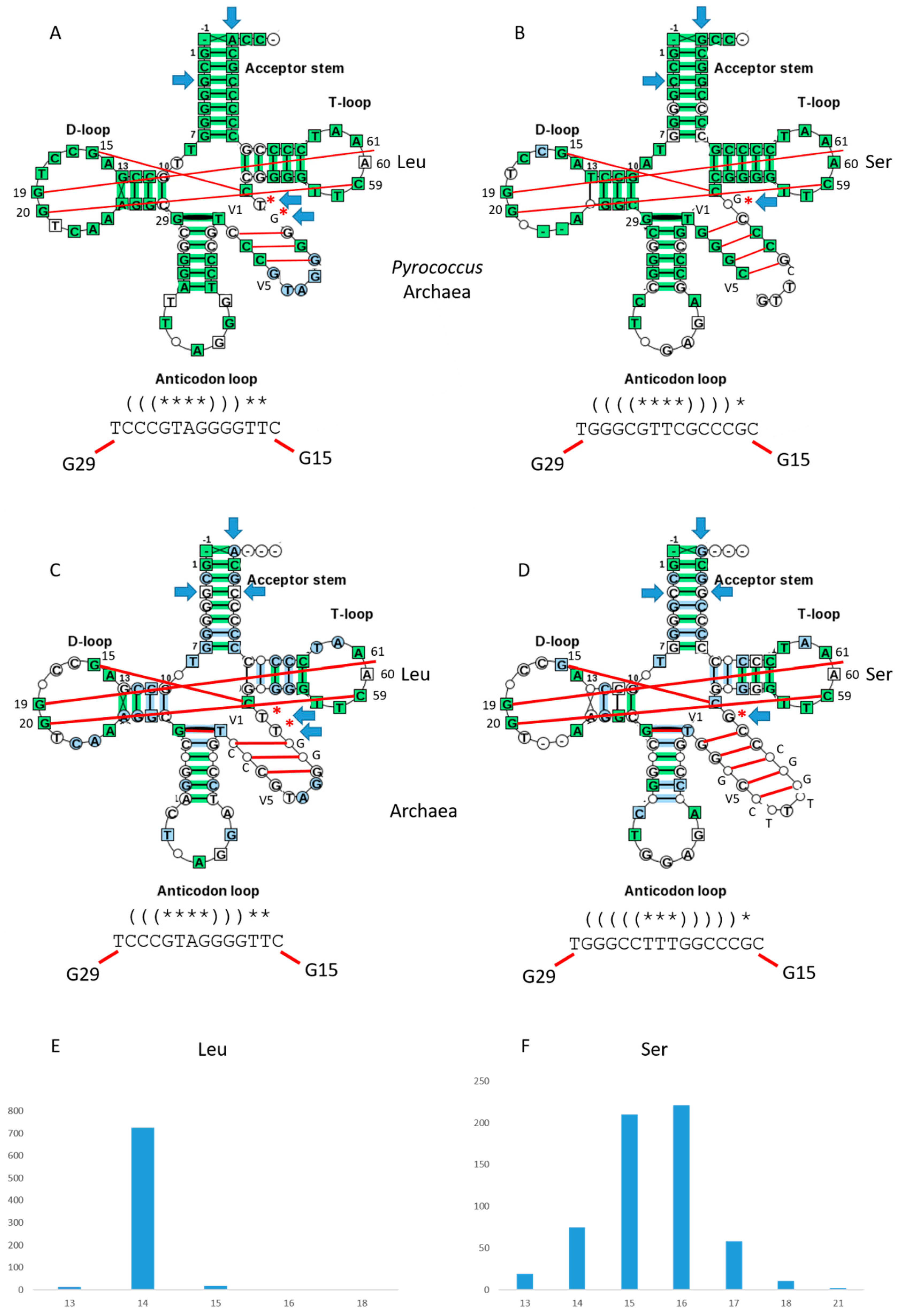
2.2. Archaeal tRNAs with Expanded V Loops
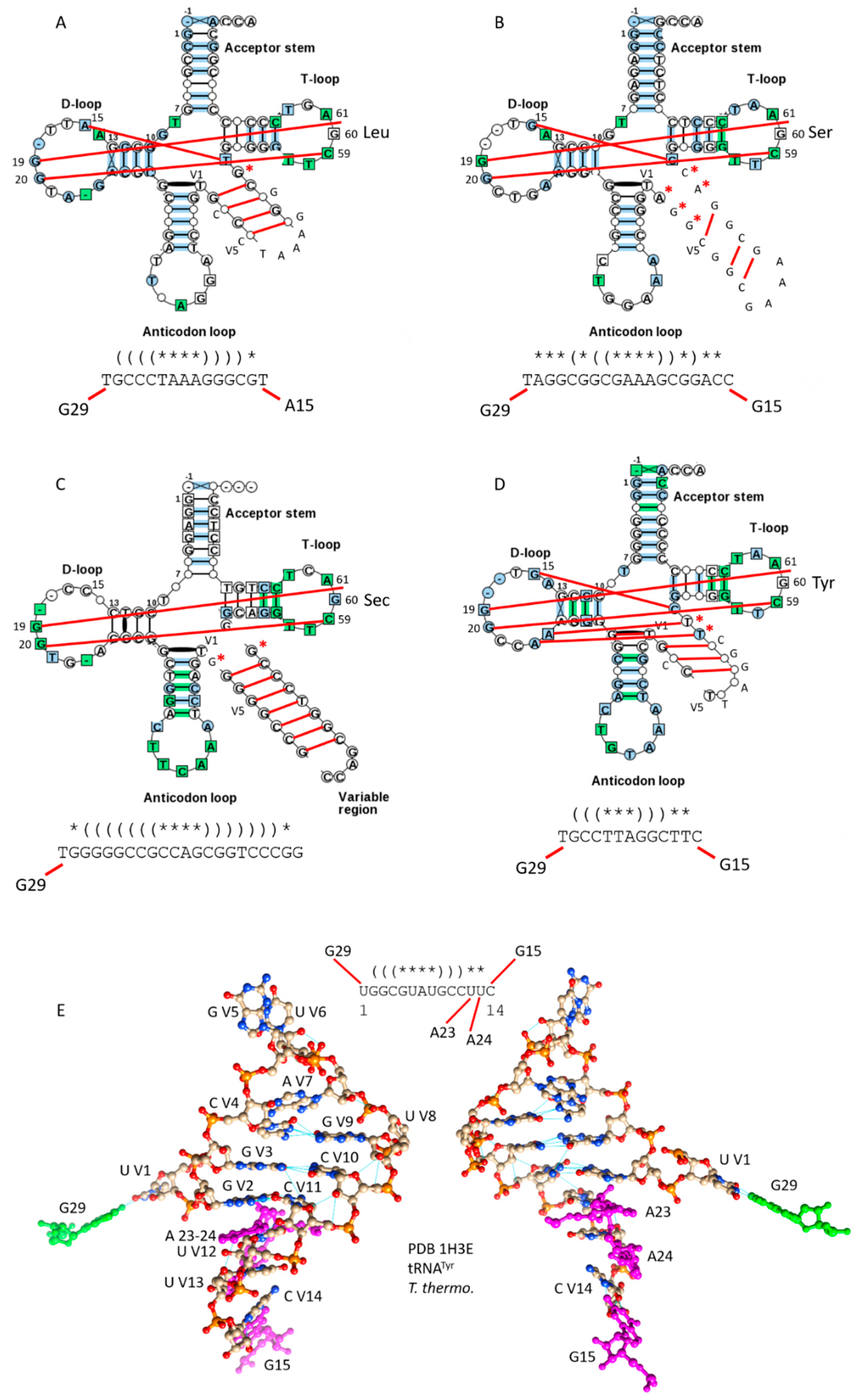
2.3. Evolution of Bacterial tRNAs with Expanded V Loops
2.4. Statistical Analyses
2.4.1. Expanded V Loops are Derived from Acceptor Stems
2.4.2. Kinship of Expanded V Loops
3. Discussion
3.1. Comparison of tRNA Evolution Models
3.2. Evolution of the Genetic Code
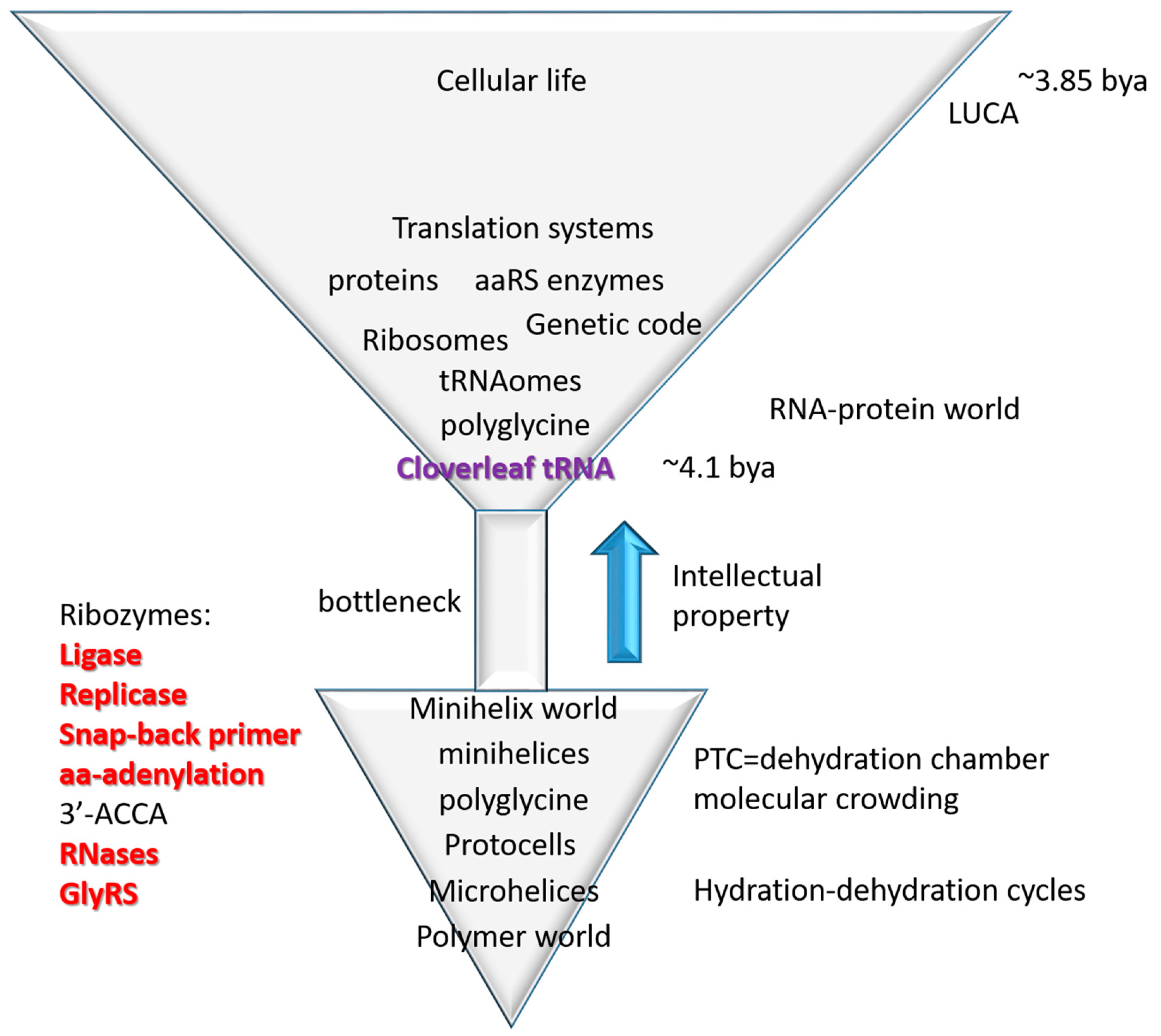
3.3. Evolution of tRNA Sequence Proceeded from Order to Chaos
3.4. The Inanimate to Animate Transition
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aaRS | Aminoacyl-tRNA synthetase (i.e., LeuRS) |
| As | Acceptor stems |
| As* | Acceptor stem remnants |
| Ac loop | Anticodon loop |
| LUCA | Last universal common (cellular) ancestor |
| T loop | T loop or TΨC loop |
| V loop | Variable loop |
References
- Pak, D.; Root-Bernstein, R.; Burton, Z.F. tRNA structure and evolution and standardization to the three nucleotide genetic code. Transcription 2017, 8, 205–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root-Bernstein, R.; Kim, Y.; Sanjay, A.; Burton, Z.F. tRNA evolution from the proto-tRNA minihelix world. Transcription 2016, 7, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhling, F.; Morl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Putz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, D159–D162. [Google Scholar] [CrossRef] [PubMed]
- Pak, D.; Du, N.; Kim, Y.; Sun, Y.; Burton, Z.F. Rooted tRNAomes and evolution of the genetic code. Transcription 2018, 9, 137–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009, 37, D93–D97. [Google Scholar] [CrossRef] [PubMed]
- Chawla, M.; Abdel-Azeim, S.; Oliva, R.; Cavallo, L. Higher order structural effects stabilizing the reverse Watson-Crick Guanine-Cytosine base pair in functional RNAs. Nucleic Acids Res. 2014, 42, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Tramontano, A.; Cavallo, L. Mg2+ binding and archaeosine modification stabilize the G15 C48 Levitt base pair in tRNAs. RNA 2007, 13, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Westhof, E.; Dumas, P.; Moras, D. Restrained refinement of two crystalline forms of yeast aspartic acid and phenylalanine transfer RNA crystals. Acta. Crystallogr. A 1988, 44 Pt 2, 112–123. [Google Scholar] [CrossRef]
- Perona, J.J.; Gruic-Sovulj, I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top Curr. Chem. 2014, 344, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Turanov, A.A.; Xu, X.M.; Carlson, B.A.; Yoo, M.H.; Gladyshev, V.N.; Hatfield, D.L. Biosynthesis of selenocysteine, the 21st amino acid in the genetic code, and a novel pathway for cysteine biosynthesis. Adv. Nutr. 2011, 2, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Chiba, S.; Sekine, S.; Yokoyama, S. Crystal structure of human selenocysteine tRNA. Nucleic Acids Res. 2009, 37, 6259–6268. [Google Scholar] [CrossRef] [PubMed]
- Yaremchuk, A.; Kriklivyi, I.; Tukalo, M.; Cusack, S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 2002, 21, 3829–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, H.S. Clues to tRNA Evolution from the Distribution of Class II tRNAs and Serine Codons in the Genetic Code. Life (Basel) 2016, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. A comparison among the models proposed to explain the origin of the tRNA molecule: A synthesis. J. Mol. Evol. 2009, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Widmann, J.; Di Giulio, M.; Yarus, M.; Knight, R. tRNA creation by hairpin duplication. J. Mol. Evol. 2005, 61, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Nagaswamy, U.; Fox, G.E. RNA ligation and the origin of tRNA. Orig. Life Evol. Biosph. 2003, 33, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Origins and Early Evolution of the tRNA Molecule. Life (Basel) 2015, 5, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Branciamore, S.; Di Giulio, M. The presence in tRNA molecule sequences of the double hairpin, an evolutionary stage through which the origin of this molecule is thought to have passed. J. Mol. Evol. 2011, 72, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, J.; Fujishima, K.; Morita, K.; Tomita, M.; Kanai, A. Disrupted tRNA gene diversity and possible evolutionary scenarios. J. Mol. Evol. 2009, 69, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Pak, D.; Burton, Z.F. Aminoacyl-tRNA synthetase proofreading, anticodon wobble preference and sectoring of the genetic code via tRNA charging errors. Transcription 2018, 9, 205–224. [Google Scholar] [CrossRef] [PubMed]
- Jose, M.V.; Govezensky, T.; Garcia, J.A.; Bobadilla, J.R. On the evolution of the standard genetic code: Vestiges of critical scale invariance from the RNA world in current prokaryote genomes. PLoS ONE 2009, 4, e4340. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F.; Eruysal, E.R.; Narendran, A.; Vare, V.Y.P.; Vangaveti, S.; Ranganathan, S.V. Celebrating wobble decoding: Half a century and still much is new. RNA Biol. 2018, 15, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Vare, V.Y.; Eruysal, E.R.; Narendran, A.; Sarachan, K.L.; Agris, P.F. Chemical and Conformational Diversity of Modified Nucleosides Affects tRNA Structure and Function. Biomolecules 2017, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F.; Vendeix, F.A.; Graham, W.D. tRNA′s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007, 366, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, M.; Surman, A.J.; Cooper, G.J.; Suarez-Marina, I.; Hosni, Z.; Lee, M.P.; Cronin, L. Formation of oligopeptides in high yield under simple programmable conditions. Nat. Commun. 2015, 6, 8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.C.; Hud, N.V.; Williams, L.D. The ribosome challenge to the RNA world. J. Mol. Evol. 2015, 80, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Illangasekare, M.; Yarus, M. Small aminoacyl transfer centers at GU within a larger RNA. RNA Biol. 2012, 9, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, R.; Lladser, M.E.; Wu, Z.; Zhang, C.; Yarus, M.; De Sterck, H.; Knight, R. Natural and artificial RNAs occupy the same restricted region of sequence space. RNA 2010, 16, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. Ahead and behind: A small, small RNA world. RNA 2015, 21, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. The meaning of a minuscule ribozyme. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2902–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, R.M.; Chumachenko, N.V.; Yarus, M. Multiple translational products from a five-nucleotide ribozyme. Proc. Natl. Acad. Sci. USA 2010, 107, 4585–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, R.M.; Illangasekare, M.; Yarus, M. Catalyzed and spontaneous reactions on ribozyme ribose. J. Am. Chem. Soc. 2011, 133, 6044–6050. [Google Scholar] [CrossRef] [PubMed]
- Chumachenko, N.V.; Novikov, Y.; Yarus, M. Rapid and simple ribozymic aminoacylation using three conserved nucleotides. J. Am. Chem. Soc. 2009, 131, 5257–5263. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, G.S.; Hendrix, D.K.; Ferrin, T.E. Nucleic acid visualization with UCSF Chimera. Nucleic Acids Res. 2006, 34, e29. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P-values against Archaeal Acceptor Stems | ||||||
| V loop | Arch LEU | Arch SER | Bact LEU | Bact SER | Bact TYR | Bact SEC |
| AVERAGE | 0.001 | 0.001 | 0.020 | 0.999 | 1.000 | 0.001 |
| P-values against Bacterial Acceptor Stems | ||||||
| V loop | Arch LEU | Arch SER | Bact LEU | Bact SER | Bact TYR | Bact SEC |
| AVERAGE | 0.001 | 0.013 | 1.000 | 0.277 | 0.020 | 0.860 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Kowiatek, B.; Opron, K.; Burton, Z.F. Type-II tRNAs and Evolution of Translation Systems and the Genetic Code. Int. J. Mol. Sci. 2018, 19, 3275. https://doi.org/10.3390/ijms19103275
Kim Y, Kowiatek B, Opron K, Burton ZF. Type-II tRNAs and Evolution of Translation Systems and the Genetic Code. International Journal of Molecular Sciences. 2018; 19(10):3275. https://doi.org/10.3390/ijms19103275
Chicago/Turabian StyleKim, Yunsoo, Bruce Kowiatek, Kristopher Opron, and Zachary F. Burton. 2018. "Type-II tRNAs and Evolution of Translation Systems and the Genetic Code" International Journal of Molecular Sciences 19, no. 10: 3275. https://doi.org/10.3390/ijms19103275
APA StyleKim, Y., Kowiatek, B., Opron, K., & Burton, Z. F. (2018). Type-II tRNAs and Evolution of Translation Systems and the Genetic Code. International Journal of Molecular Sciences, 19(10), 3275. https://doi.org/10.3390/ijms19103275