Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response


Abstract
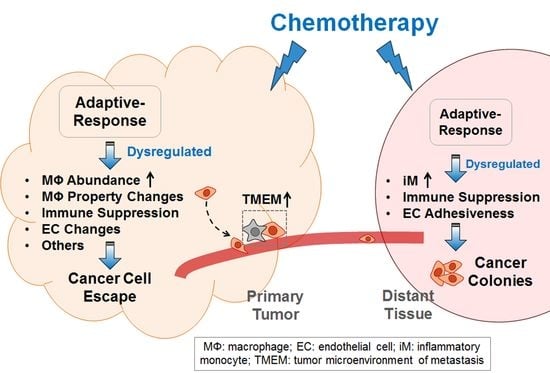
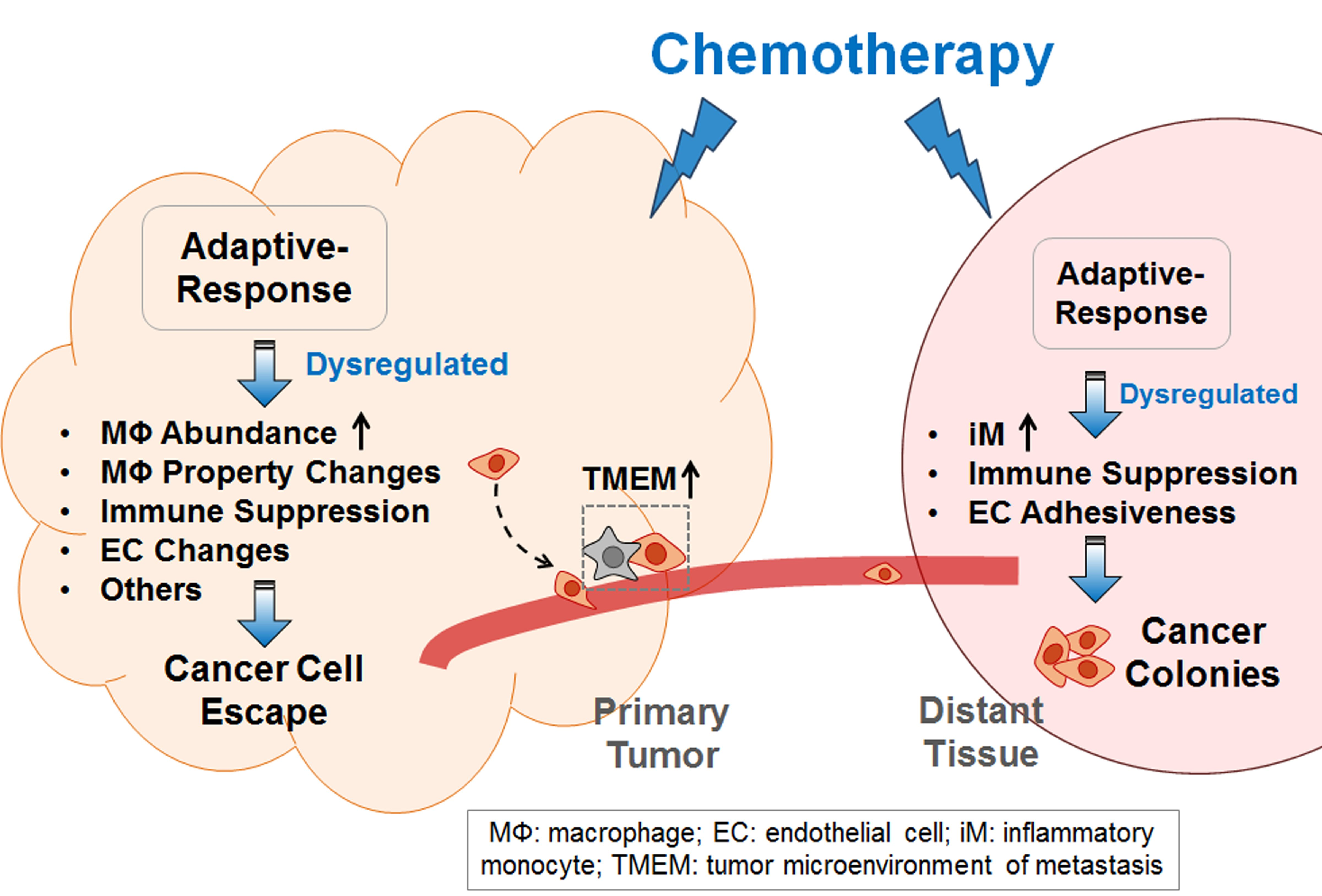
:
1. The Double-Edged Sword of Chemotherapy-Findings from Mouse Models
1.1. The Paradox of Chemotherapy
1.2. The Pro-Cancer Effect of Chemotherapy—Chemo-Resistance versus Chemo-Exacerbation
1.2.1. Chemo-Resistance: Chemotherapy Counteracts Its Own Efficacy
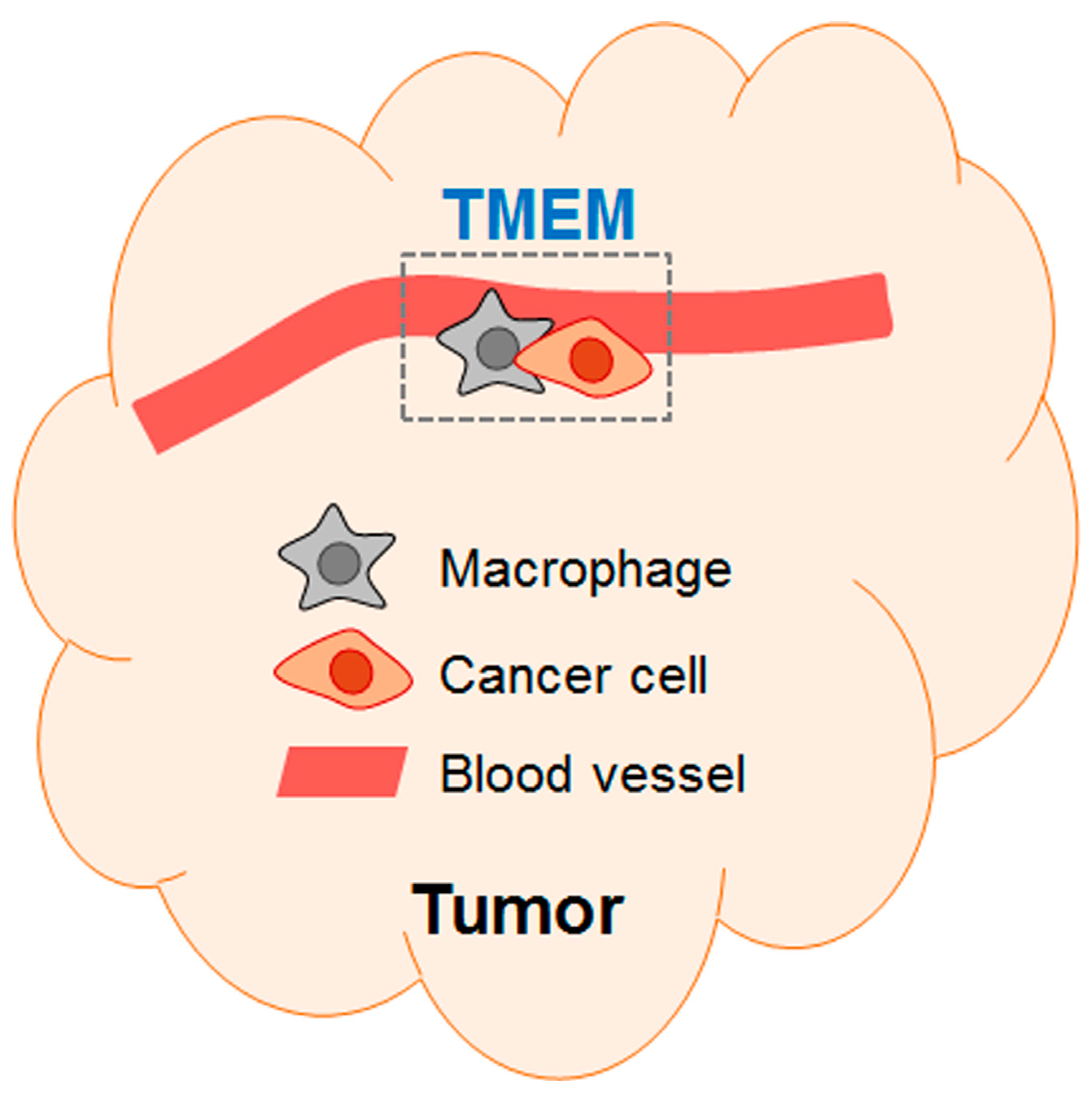
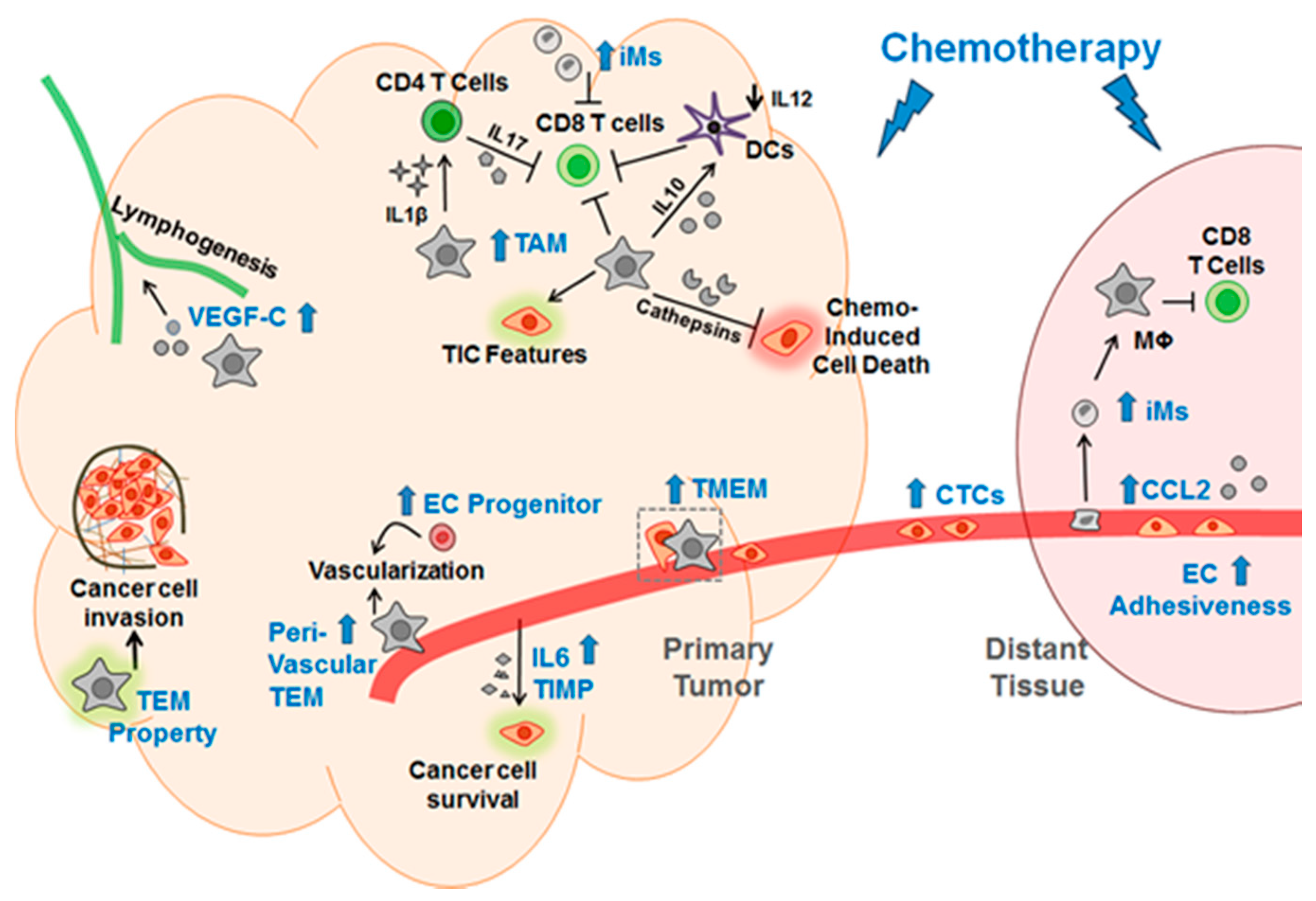
1.2.2. Chemo-Exacerbation: Chemotherapy Exacerbates Metastasis
1.3. Pre-Metastatic versus Metastatic Niche
1.4. The Culprit in the Host—the Key Non-Cancer Cells Contributing to the Pro-Cancer Effect of Chemotherapy
1.4.1. Myeloid Cells
1.4.2. Endothelial Cells
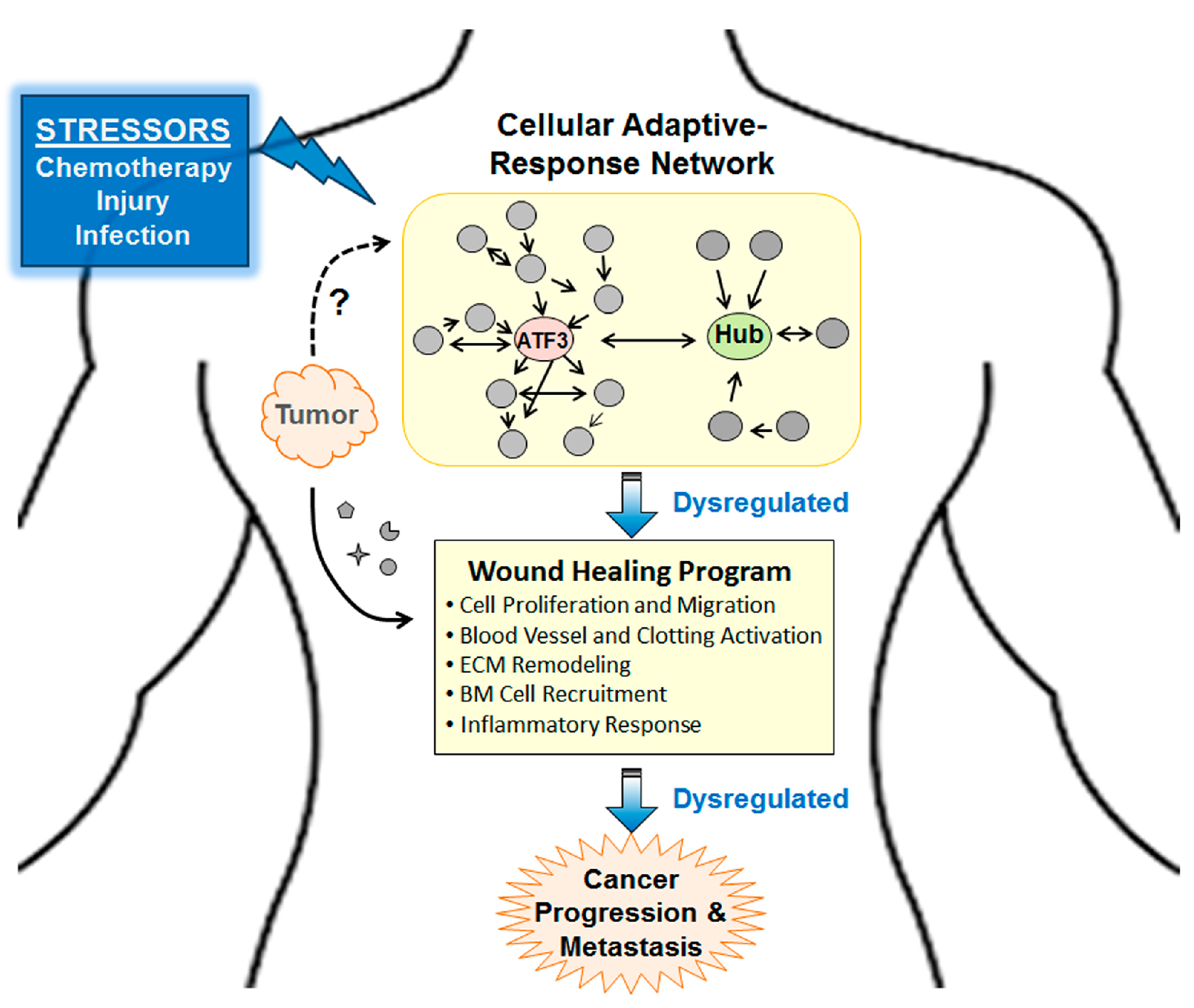
2. Explaining the Pro-Cancer Effect of Chemotherapy from the Perspective of Adaptive-Response
3. The Relevance of the above Findings to Human Breast Cancer
4. Should Findings from Mouse Models Affect Clinical Practices?
4.1. Neoadjuvant (Pre-Operative) Chemotherapy
4.2. Adjuvant (Post-Operative) Chemotherapy
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF3 | Activating transcription factor 3 |
| CD | Cluster of differentiation |
| iM | Inflammatory monocyte |
| TAM | Tumor associated macrophage |
| TEM | Tie2-expressing macrophage |
| TIC | Tumor initiation cell |
| TLR | Toll-like receptor |
| TMEM | Tumor microenvironment metastasis |
| WT | Wild type |
References
- Wang, A.C.; Su, Q.B.; Wu, F.X.; Zhang, X.L.; Liu, P.S. Role of TLR4 for paclitaxel chemotherapy in human epithelial ovarian cancer cells. Eur. J. Clin. Investig. 2009, 39, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, M.; Elia, L.; Price, J.H.; Heynen-Genel, S.; Courtneidge, S.A. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci. Signal. 2011, 4, ra49. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhou, X.; Yang, J.J.; Liu, X.; Zhao, X.H.; Wang, Q.X.; Han, L.; Song, X.; Zhu, Z.Y.; Tian, W.P.; et al. AC1MMYR2 impairs high dose paclitaxel-induced tumor metastasis by targeting miR-21/CDK5 axis. Cancer Lett. 2015, 362, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Hemann, M.T. Chemotherapeutic resistance: Surviving stressful situations. Cancer Res. 2011, 71, 5062–5066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiao, S.L.; Ganesan, A.P.; Rugo, H.S.; Coussens, L.M. Immune microenvironments in solid tumors: New targets for therapy. Genes Dev. 2011, 25, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2002, 5, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Johansson, M.; Coussens, L.M. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2008, 27, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2012, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, E.S.; Askautrud, H.A.; Kees, T.; Park, J.H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.X.; Qiu, J.; et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis: mechanisms and translational opportunities. Clin. Exp. Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Volk-Draper, L.; Hall, K.; Griggs, C.; Rajput, S.; Kohio, P.; DeNardo, D.; Ran, S. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 2014, 74, 5421–5434. [Google Scholar] [CrossRef] [PubMed]
- Daenen, L.G.; Roodhart, J.M.; van Amersfoort, M.; Dehnad, M.; Roessingh, W.; Ulfman, L.H.; Derksen, P.W.; Voest, E.E. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011, 71, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
- Gingis-Velitski, S.; Loven, D.; Benayoun, L.; Munster, M.; Bril, R.; Voloshin, T.; Alishekevitz, D.; Bertolini, F.; Shaked, Y. Host response to short-term, single-agent chemotherapy induces matrix metalloproteinase-9 expression and accelerates metastasis in mice. Cancer Res. 2011, 71, 6986–6996. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Liao, J.; Berry, J.E.; Li, X.; Koh, A.J.; Michalski, M.E.; Eber, M.R.; Soki, F.N.; Sadler, D.; Sud, S.; et al. Cyclophosphamide creates a receptive microenvironment for prostate cancer skeletal metastasis. Cancer Res. 2012, 72, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Alishekevitz, D.; Gingis-Velitski, S.; Kaidar-Person, O.; Gutter-Kapon, L.; Scherer, S.D.; Raviv, Z.; Merquiol, E.; Ben-Nun, Y.; Miller, V.; Rachman-Tzemah, C.; et al. Macrophage-Induced Lymphangiogenesis and Metastasis following Paclitaxel Chemotherapy Is Regulated by VEGFR3. Cell Rep. 2016, 17, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Jalgaonkar, S.P.; Middleton, J.D.; Hai, T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E7159–E7168. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.D.; Sica, G.L.; Liu, Y.F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor microenvironment of metastasis in human breast carcinoma: A potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1889, 8, 98–101. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.C.; Lu, X.; Yu, M.; Lemos, H.; Huang, L.; Chandler, P.; Liu, K.; Walters, M.; Krasinski, A.; Mack, M.; et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014, 74, 3441–3453. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Shaked, Y.; Henke, E.; Roodhart, J.M.; Mancuso, P.; Langenberg, M.H.; Colleoni, M.; Daenen, L.G.; Man, S.; Xu, P.; Emmenegger, U.; et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell 2008, 14, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Roodhart, J.M.; Langenberg, M.H.; Vermaat, J.S.; Lolkema, M.P.; Baars, A.; Giles, R.H.; Witteveen, E.O.; Voest, E.E. Late release of circulating endothelial cells and endothelial progenitor cells after chemotherapy predicts response and survival in cancer patients. Neoplasia 2010, 12, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. ATF3 and stress responses. Gene Expr. 1999, 7, 321–335. [Google Scholar] [PubMed]
- Hai, T.; Wolford, C.C.; Chang, Y.S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.H.; Murray, G.I.; Stevenson, D.A.; Haites, N.E.; Cassidy, J.; McLeod, H.L. Comparative genomic hybridization and chromosomal instability in solid tumours. Br. J. Cancer 1999, 80, 862–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; DeWille, J.; Hai, T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 2008, 27, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Arantes, S.; Yan, L.; Kiguchi, K.; McArthur, M.J.; Sahin, A.; Thames, H.D.; Aldaz, C.M.; Macleod, M.C. The transcription factor ATF3 acts as an oncogene in mouse mammary tumorigenesis. BMC Cancer 2008, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wolford, C.C.; Chang, Y.S.; McConoughey, S.J.; Ramsey, S.A.; Aderem, A.; Hai, T. ATF3, an adaptive-response gene, enhances TGF{beta} signaling and cancer-initiating cell features in breast cancer cells. J. Cell Sci. 2010, 123, 3558–3565. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Della Coletta, L.; Powell, K.L.; Shen, J.; Thames, H.; Aldaz, C.M.; MacLeod, M.C. Activation of the canonical Wnt/beta-catenin pathway in ATF3-induced mammary tumors. PLoS ONE 2011, 6, e16515. [Google Scholar]
- Wolford, C.C.; McConoughey, S.J.; Jalgaonkar, S.P.; Leon, M.; Merchant, A.S.; Dominick, J.L.; Yin, X.; Chang, Y.; Zmuda, E.J.; Toole, S.A.; et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J. Clin. Investig. 2013, 123, 2893–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.R.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10, eaan3464. [Google Scholar] [CrossRef] [PubMed]
- Rachman-Tzemah, C.; Zaffryar-Eilot, S.; Grossman, M.; Ribero, D.; Timaner, M.; Maki, J.M.; Myllyharju, J.; Bertolini, F.; Hershkovitz, D.; Sagi, I.; et al. Blocking Surgically Induced Lysyl Oxidase Activity Reduces the Risk of Lung Metastases. Cell Rep. 2017, 19, 774–784. [Google Scholar] [CrossRef] [PubMed]
- El Saghir, N.S.; Elhajj, I.I.; Geara, F.B.; Hourani, M.H. Trauma-associated growth of suspected dormant micrometastasis. BMC Cancer 2005, 5, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleeman, J.P. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012, 31, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, J.C.; Wang, J.H.; Smith, M.J.; Bouchier-Hayes, D.; Cotter, T.G.; Redmond, H.P. Excisional surgery for cancer cure: therapy at a cost. Lancet. Oncol. 2003, 4, 760–768. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Emmenegger, U.; Kerbel, R.S. Cancer: Chemotherapy counteracted. Nature 2010, 468, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr266. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The seed and soil hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Engenhart-Cabillic, R.; Daniilidis, K.; Rose, F.; An, H.X. Metastasis: The seed and soil theory gains identity. Cancer Metastasis Rev. 2007, 26, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Kaplan, R.N.; Port, E.R.; Lyden, D. Priming the “soil” for breast cancer metastasis: The pre-metastatic niche. Breast Dis. 2006, 26, 65–74. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConkey, D.J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Gralow, J.R.; Burstein, H.J.; Wood, W.; Hortobagyi, G.N.; Gianni, L.; von Minckwitz, G.; Buzdar, A.U.; Smith, I.E.; Symmans, W.F.; Singh, B.; et al. Preoperative therapy in invasive breast cancer: Pathologic assessment and systemic therapy issues in operable disease. J. Clin. Oncol. 2008, 26, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, P.; Anderson, S.J.; Bear, H.D.; Geyer, C.E.; Kahlenberg, M.S.; Robidoux, A.; Margolese, R.G.; Hoehn, J.L.; Vogel, V.G.; Dakhil, S.R.; et al. Preoperative chemotherapy: Updates of National Surgical Adjuvant Breast and Bowel Project Protocols B-18 and B-27. J. Clin. Oncol. 2008, 26, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, G.; Brusamolino, E.; Valagussa, P.; Rossi, A.; Brugnatelli, L.; Brambilla, C.; De Lena, M.; Tancini, G.; Bajetta, E.; Musumeci, R.; et al. Combination chemotherapy as an adjuvant treatment in operable breast cancer. N. Engl. J. Med. 1976, 294, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Sarah, D.; Christina, D.; Paul, M. The early breast caner trialists’ collaborative group: A brief history of results to date. In Celebrating Statistics: Papers in Honour of Sir David Cox on His 80th Birthday; Davison, A.C., Dodge, Y., Wermuth, N.S., Eds.; Oxford University Press: Oxford, UK, 2005; pp. 185–198. [Google Scholar]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Cook, A.D.; Tak, P.P. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat. Rev. Drug Discov. 2016, 16, 53–70. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Macrophage Actions | Some Key Points | References |
|---|---|---|
| (a) Alter cancer cell behavior (or activity) |
| Shree et al., 2011 [20]. |
| Mitchem et al., 2012 [18] | |
| Chang et al., 2017 [27] | |
| (b) Suppress cytotoxic CD8+ T cells |
| Mitchem et al., 2012 [18] |
| DeNardo et al., 2011 and Ruffell et al., 2014 [17,35] | |
| Bruchard et al., 2013 [36] | |
| Ding et al., 2014 [37] | |
| (c) Alter blood or lymphatic vessels |
| Hughes et al., 2015 [38] |
| Chang et al., 2017 and Karagiannis et al., 2017 [27,28] | |
| Alishekevitz et al., 2016 [26] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Middleton, J.D.; Stover, D.G.; Hai, T. Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. Int. J. Mol. Sci. 2018, 19, 3333. https://doi.org/10.3390/ijms19113333
Middleton JD, Stover DG, Hai T. Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. International Journal of Molecular Sciences. 2018; 19(11):3333. https://doi.org/10.3390/ijms19113333
Chicago/Turabian StyleMiddleton, Justin D., Daniel G. Stover, and Tsonwin Hai. 2018. "Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response" International Journal of Molecular Sciences 19, no. 11: 3333. https://doi.org/10.3390/ijms19113333
APA StyleMiddleton, J. D., Stover, D. G., & Hai, T. (2018). Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. International Journal of Molecular Sciences, 19(11), 3333. https://doi.org/10.3390/ijms19113333