Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Coarse-Grained Protein Modeling
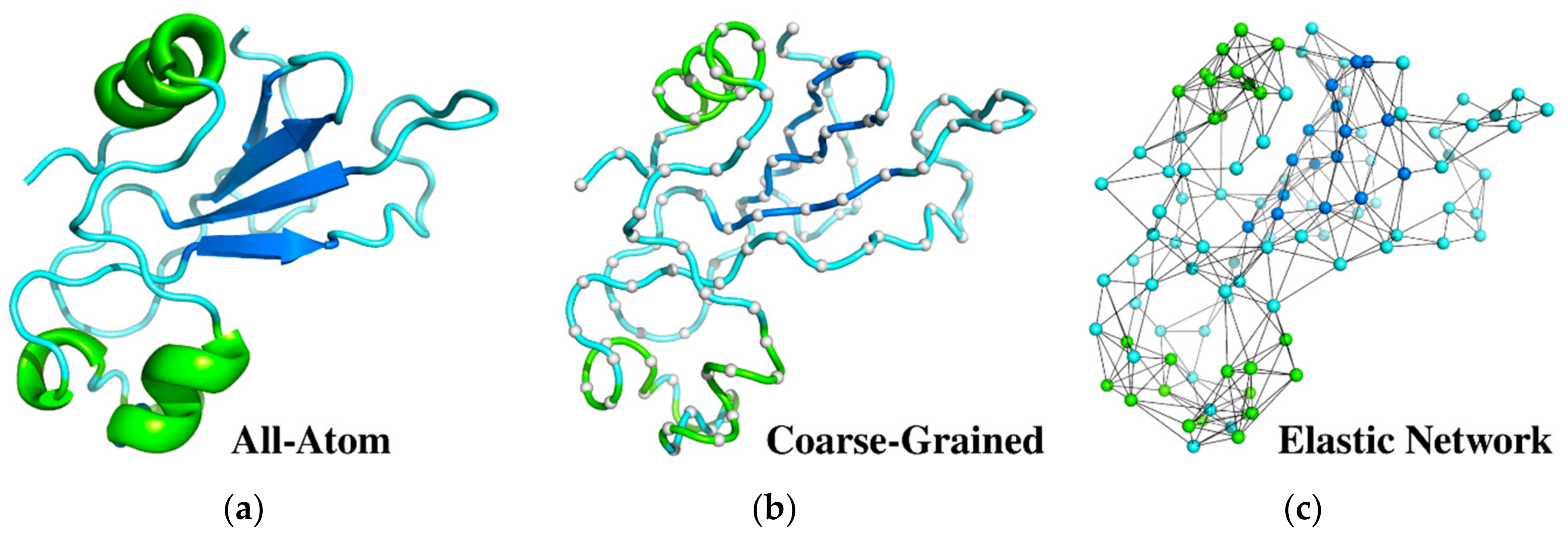
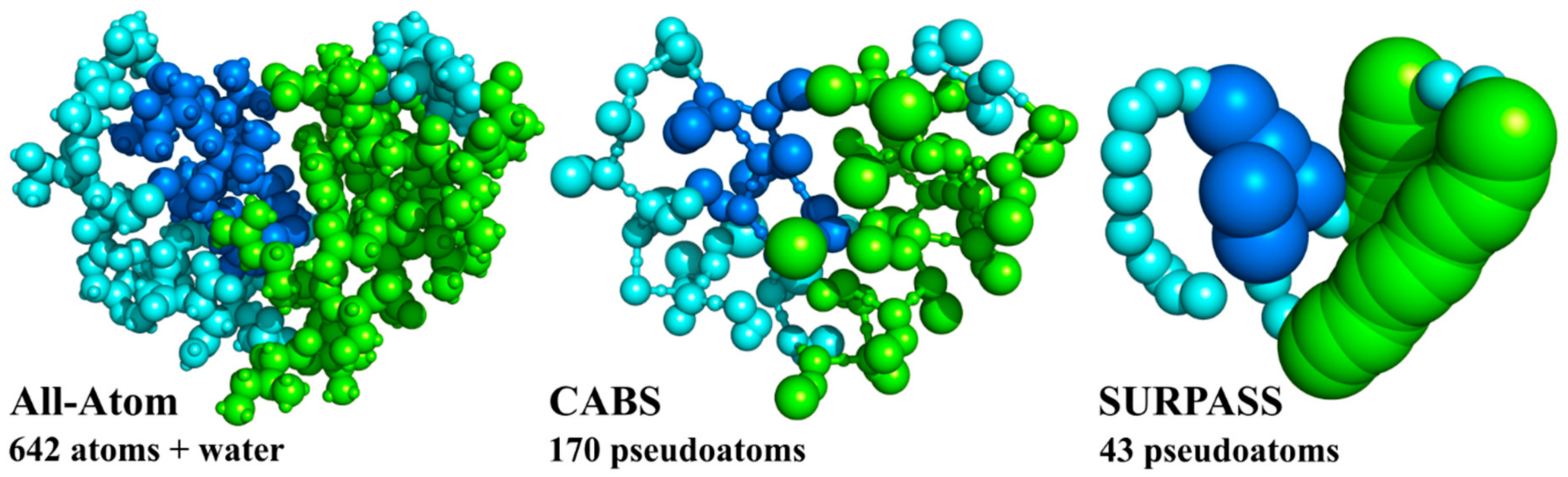
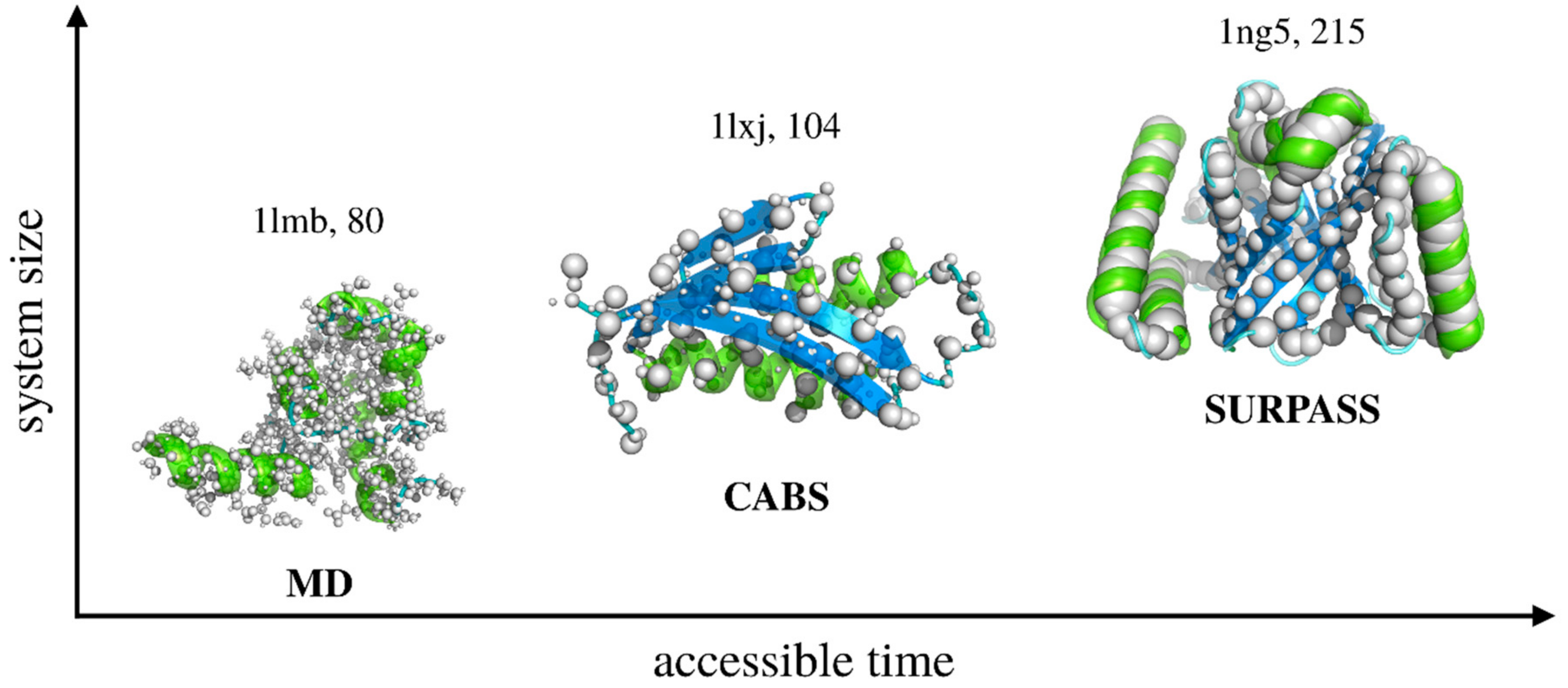
2.1. From All-Atom to Coarse-Grained Modeling
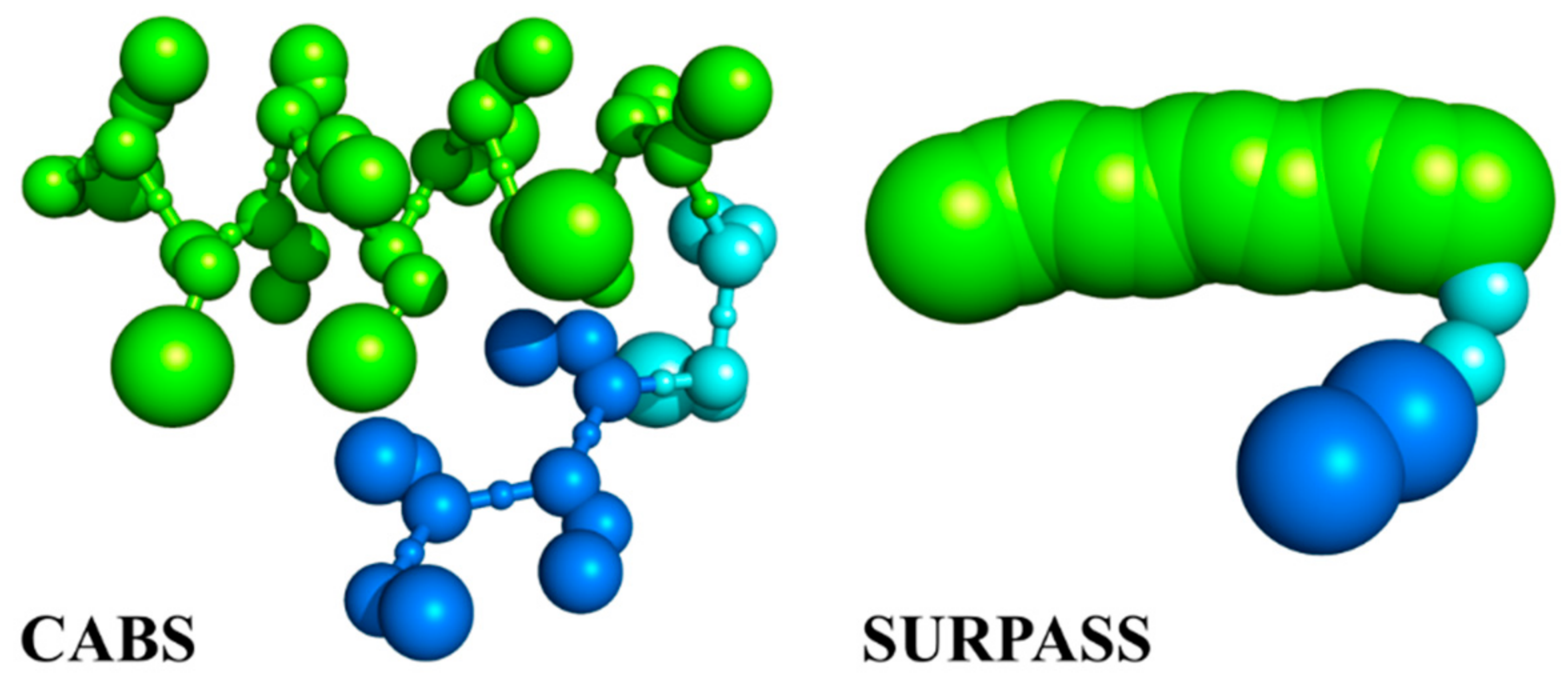
2.2. Coarse-Grained CABS Model
2.3. Coarse-Grained SURPASS Model
2.4. Elastic Network Models
3. Applications of Coarse-Grained Modeling: ENM and CG Monte Carlo Simulations
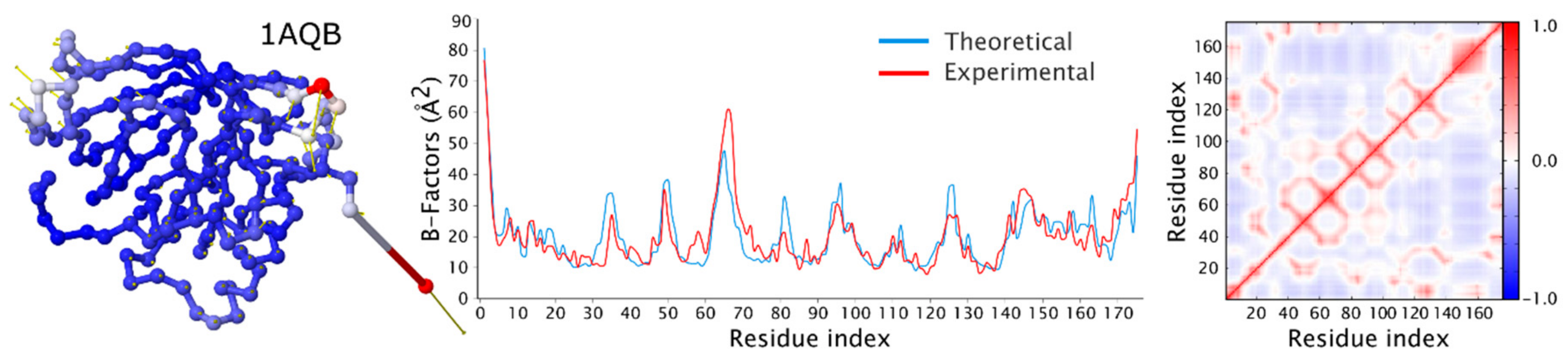
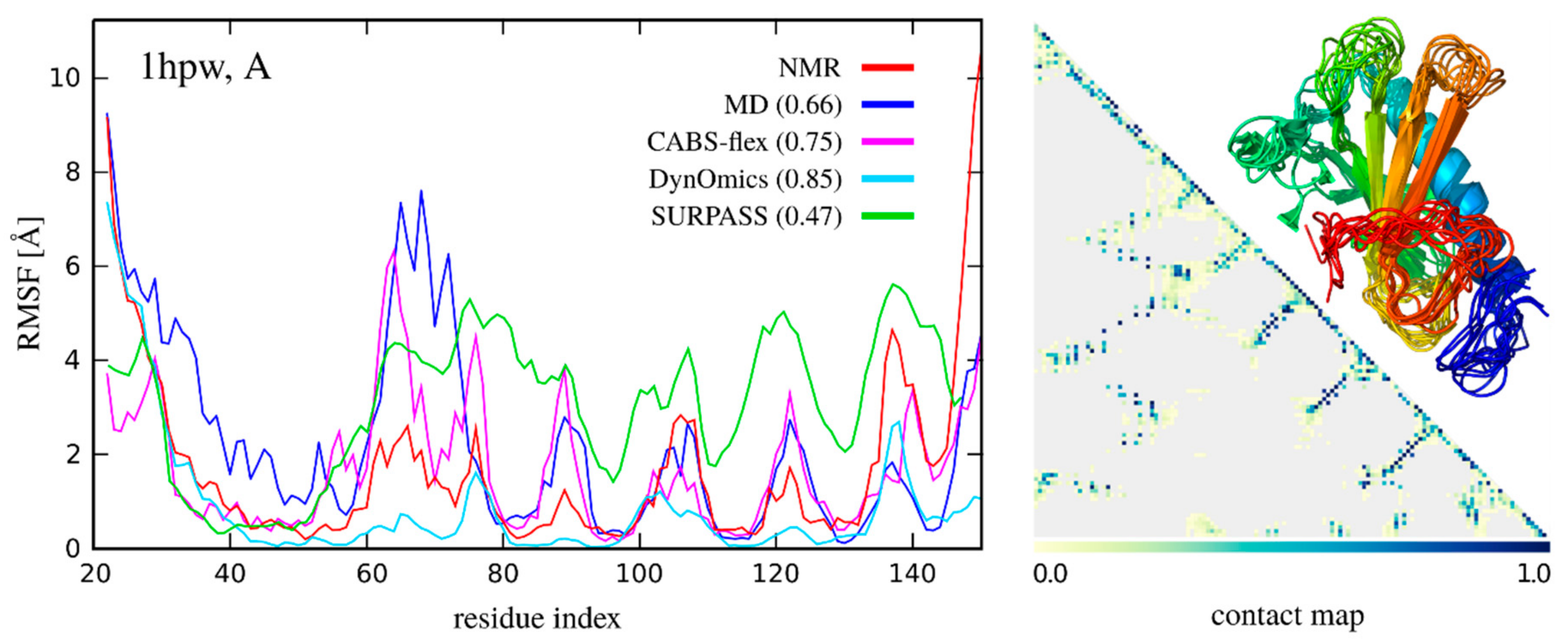
3.1. Modeling of the Structural Flexibility of Folded Proteins
- NMR ensemble: data calculated using 10 models deposited in the PDB code: 1hpw.
- MD all-atom simulation: data obtained using a 10 nanosecond trajectory with an AMBER8.0 force field taken from the MoDEL database of MD trajectories [38]; RMSF was calculated for the entire trajectory consisting of 10,000 models.
- CG simulation using the CABS model: data obtained using the CABS-flex 2.0 web server [39]; results calculated using the default server settings; RMSF was calculated for the set of 10 representative models (obtained by a cluster analysis of 10,000 snapshots) from the simulation trajectory.
- CG simulation using the SURPASS model: data obtained with the following SURPASS [25] settings: isothermal MC simulation in low reduced temperature (T = 0.2), 10,000 MC steps, 1 and 3-bead motions; RMSF was calculated for the entire trajectory of 100 models.
- ENM modeling: data obtained using the DynOmics web server [85,95] that integrates two ENM methods: the GNM and the ANM, calculated using default server settings; real time calculation: <1 min; RMSF was calculated for the set of 20 models based on the 10 slowest modes (2 models per mode for extreme positions during movement); the amplitude of motion along each mode was chosen so that the RMSD (root-mean-square deviation of atomic positions) between the models was less than 2Å; all models were generated using the ‘Molecular Motions—Animation’ option available on the results page of the DynOmics server.
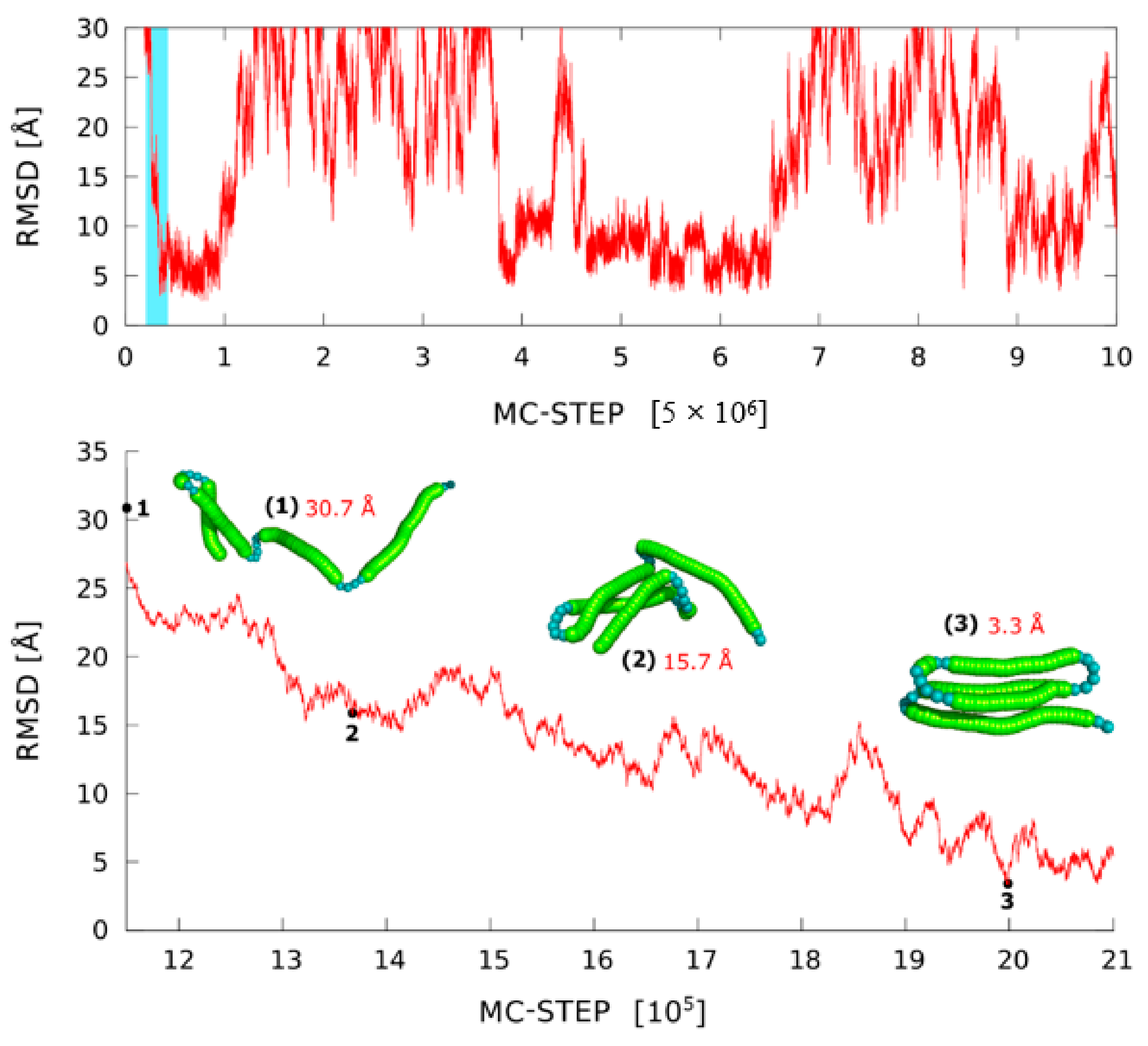
3.2. Modeling Large-Scale Structural Transitions
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MD | molecular dynamics |
| CG | coarse-grained |
| ENM | elastic network model |
| NMA | normal mode analysis |
| MC | Monte Carlo |
| AA | all-atom |
| CABS | Cα, Cβ, Side chain model |
| SURPASS | Single United Residue per Pre-averaged Secondary Structure fragment |
| REMC | replica exchange Monte Carlo |
| PCA | principle component analysis |
| PCs | principle components |
| GNM | Gaussian network model |
| PDB | Protein Data Bank |
| ANM | anisotropic network model |
| GPCR | G protein-coupled receptor |
| RMSF | root mean square fluctuations |
| RMSD | root-mean-square deviation of atomic positions |
| NMR | nuclear magnetic resonance |
| UNRES | united residue model |
| GEN | generalized elastic network model |
| AFM | atomic force microscope |
| CHARMM | Chemistry at HARvard Macromolecular Mechanics |
References
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, H.X. Protein allostery and conformational dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Cossins, B.P.; Lawson, A.D.G.; Shi, J. Computational exploration of conformational transitions in protein drug targets. Methods Mol. Biol. 2018, 1762, 339–365. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Devaurs, D.; Kavraki, L.E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discov. 2015, 10, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein-peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.C.; Weinreich, T.M.; Piana, S.; Shaw, D.E. Demonstrating an order-of-magnitude sampling enhancement in molecular dynamics simulations of complex protein systems. J. Chem. Theory Comput. 2016, 12, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed]
- Kouza, M.; Hu, C.K.; Li, M.S.; Kolinski, A. A structure-based model fails to probe the mechanical unfolding pathways of the titin I27 domain. J. Chem. Phys. 2013, 139, 065103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hills, R.D.; Brooks, C.L. Insights from coarse-grained Gō models for protein folding and dynamics. Int. J. Mol. Sci. 2009, 10, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Orozco, M. A theoretical view of protein dynamics. Chem. Soc. Rev. 2014, 43, 5051–5066. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Rader, A.J. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005, 15, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Blanco, J.R.; Chacón, P. New generation of elastic network models. Curr. Opin. Struct. Biol. 2016, 37, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, M. Birth and future of multiscale modeling for macromolecular systems (Nobel Lecture). Angew. Chemi. Int. Ed. 2014, 53, 10006–10018. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A. Multiscale modeling of biological functions: From enzymes to molecular machines (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2014, 53, 10020–10031. [Google Scholar] [CrossRef] [PubMed]
- Stumpff-Kane, A.W.; Maksimiak, K.; Lee, M.S.; Feig, M. Sampling of near-native protein conformations during protein structure refinement using a coarse-grained model, normal modes, and molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2008, 70, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Kolinski, A. Toward more efficient simulations of slow processes in large biomolecular systems: Comment on “Ligand diffusion in proteins via enhanced sampling in molecular dynamics” by Jakub Rydzewski and Wieslaw Nowak. Phys. Life Rev. 2017, 22–23, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V. Minimalist models for proteins: A comparative analysis. Q. Rev. Biophys. 2010, 43, 333–371. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol. 2005, 15, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Baranowski, M.; Czaplewski, C.; Gołaś, E.; He, Y.; Jagieła, D.; Krupa, P.; Maciejczyk, M.; Makowski, M.; Mozolewska, M.A.; et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model. 2014, 20, 2306–2321. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Gopal, S.M.; Cheng, Y.M.; Predeus, A.; Feig, M. PRIMO: A transferable coarse-grained force field for proteins. J. Chem. Theory Comput. 2013, 9, 3769–3788. [Google Scholar] [CrossRef] [PubMed]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar]
- Dawid, A.E.; Gront, D.; Kolinski, A. SURPASS low-resolution coarse-grained protein modeling. J. Chem. Theory Comput. 2017, 13, 5766–5779. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Kolinski, A. One-dimensional structural properties of proteins in the coarse-grained cabs model. Methods Mol. Biol. 2017, 1484, 83–113. [Google Scholar] [CrossRef] [PubMed]
- Godzik, A.; Kolinski, A.; Skolnick, J. Lattice representations of globular proteins: How good are they? J. Comput. Chem. 1993, 14, 1194–1202. [Google Scholar] [CrossRef]
- Kmiecik, S.; Kolinski, A. Characterization of protein-folding pathways by reduced-space modeling. Proc. Natl. Acad. Sci. USA 2007, 104, 12330–12335. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Kurcinski, M.; Rutkowska, A.; Gront, D.; Kolinski, A. Denatured proteins and early folding intermediates simulated in a reduced conformational space. Acta Biochim. Pol. 2006, 53, 131–144. [Google Scholar] [PubMed]
- Kmiecik, S.; Kolinski, A. Folding pathway of the B1 domain of protein G explored by multiscale modeling. Biophys. J. 2008, 94, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kouza, M.; Kolinski, A. From coarse-grained to atomic-level characterization of protein dynamics: Transition state for the folding of B domain of protein A. J. Phys. Chem. B 2012, 116, 7026–7032. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Kolinski, A. simulation of chaperonin effect on protein folding: A shift from nucleation-condensation to framework mechanism. J. Am. Chem. Soc. 2011, 133, 10283–10289. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Wabik, J.; Kolinski, M.; Kouza, M.; Kolinski, A. Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes; Coarse-Grained Modeling of Protein Dynamics; Springer: Berlin, Germany, 2014; pp. 55–79. [Google Scholar]
- Kurcinski, M.; Kolinski, A.; Kmiecik, S. Mechanism of folding and binding of an intrinsically disordered protein as revealed by ab initio simulations. J. Chem. Theory Comput. 2014, 10, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Debinski, A.; Paczkowska, M.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. Protein-peptide molecular docking with large-scale conformational changes: The p53-MDM2 interaction. Sci. Rep. 2016, 6, 37532. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Orozco, M.; Kolinski, A.; Kmiecik, S. Consistent view of protein fluctuations from all-atom molecular dynamics and coarse-grained dynamics with knowledge-based force-field. J. Chem. Theory Comput. 2013, 9, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Kolinski, A.; Kmiecik, S. CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013, 41, W427–W431. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Kolinski, A.; Kmiecik, S. CABS-flex predictions of protein flexibility compared with NMR ensembles. Bioinformatics 2014, 30, 2150–2154. [Google Scholar] [CrossRef] [PubMed]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Oleniecki, T.; Ciemny, P.M.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Pulawski, W.; Jamroz, M.; Kolinski, M.; Kolinski, A.; Kmiecik, S. Coarse-grained simulations of membrane insertion and folding of small helical proteins using the CABS model. J. Chem. Inf. Model. 2016, 56, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, M.; Jamroz, M.; Kmiecik, S.; Kolinski, A. CABS-fold: Server for the de novo and consensus-based prediction of protein structure. Nucleic Acids Res. 2013, 41, W406–W411. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Jamroz, M.; Kolinski, M. Structure prediction of the second extracellular loop in G-protein-coupled receptors. Biophys. J. 2014, 106, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Kolinski, A.; Bujnicki, J.M. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins Struct. Funct. Genet. 2005, 61, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, M.; Kurcinski, M.; Kouza, M.; Wieteska, L.; Debinski, A.; Kolinski, A.; Kmiecik, S. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016, 93, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Kurcinski, M.; Kozak, K.; Kolinski, A.; Kmiecik, S. Highly flexible protein-peptide docking using cabs-dock. Methods Mol. Biol. 2017, 1561, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Kurcinski, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. Modeling EphB4-EphrinB2 protein-protein interaction using flexible docking of a short linear motif. Biomed. Eng. Online 2017, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Blaszczyk, M.; Ciemny, M.P.; Kolinski, A.; Kmiecik, S. A protocol for CABS-dock protein-peptide docking driven by side-chain contact information. Biomed. Eng. Online 2017, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Kolinski, A.; Kmiecik, S. Protocols for efficient simulations of long-time protein dynamics using coarse-grained CABS model. Methods Mol. Biol. 2014, 1137, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Dawid, A.E.; Gront, D.; Kolinski, A. Coarse-grained modeling of the interplay between secondary structure propensities and protein fold assembly. J. Chem. Theory Comput. 2018, 14, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Feig, M.; Rotkiewicz, P.; Kolinski, A.; Skolnick, J.; Brooks, C.L. Accurate reconstruction of all-atom protein representations from side-chain-based low-resolution models. Proteins Struct. Funct. Genet. 2000, 41, 86–97. [Google Scholar] [CrossRef]
- Yang, L.; Song, G.; Jernigan, R.L. How well can we understand large-scale protein motions using normal modes of elastic network models? Biophys. J. 2007, 93, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Ma, J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure 2005, 13, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal mode analysis of biomolecular structures: Functional mechanisms of membrane proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.; De Groot, B.L. Normal modes and essential dynamics. Methods Mol. Biol. 2008, 443, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Toussi, C.A.; Soheilifard, R. A better prediction of conformational changes of proteins using minimally connected network models. Phys. Biol. 2017, 13, 066013. [Google Scholar] [CrossRef] [PubMed]
- Fuglebakk, E.; Tiwari, S.P.; Reuter, N. Comparing the intrinsic dynamics of multiple protein structures using elastic network models. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 911–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Lee, B.H.; Kim, M.K. Robust elastic network model: A general modeling for precise understanding of protein dynamics. J. Struct. Biol. 2015, 190, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Van Wynsberghe, A.W.; Cui, Q. Interpreting correlated motions using normal mode analysis. Structure 2006, 14, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Rueda, M.; Chacón, P.; Orozco, M. Thorough validation of protein normal mode analysis: A comparative study with essential dynamics. Structure 2007, 15, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Go, N.; Noguti, T.; Nishikawa, T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. USA 1983, 80, 3696–3700. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.; Brooks, B.; Karplus, M.; Karplus, M. Harmonic dynamics of proteins: Normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc Natl Acad Sci USA 1983, 80, 6571–6575. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, J. The role of shape in determining molecular motions. Biophys. J. 2005, 89, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Howe, P.W. Principal components analysis of protein structure ensembles calculated using NMR data. J. Biomol. NMR 2001, 20, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Song, G.; Carriquiry, A.; Jernigan, R.L. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure 2008, 16, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, A.M.; Kirys, T.; Tuzikov, A.V.; Vakser, I.A. Structure fluctuations and conformational changes in protein binding. J. Bioinform. Comput. Biol. 2012, 10, 1241002. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Bahar, I.; Wallqvist, A.; Covell, D.G.; Jernigan, R.L. Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry 1998, 37, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Eyal, E.; Yang, L.-W.; Bahar, I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics 2006, 22, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Doruker, P.; Jernigan, R.L.; Bahar, I. Dynamics of large proteins through hierarchical levels of coarse-grained structures. J. Comput. Chem. 2002, 23, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Kurkcuoglu, O.; Jernigan, R.L.; Doruker, P. Mixed levels of coarse-graining of large proteins using elastic network model succeeds in extracting the slowest motions. Polymer 2004, 45, 649–657. [Google Scholar] [CrossRef]
- Mishra, S.K.; Jernigan, R.L. Protein dynamic communities from elastic network models align closely to the communities defined by molecular dynamics. PLoS ONE 2018, 13, e0199225. [Google Scholar] [CrossRef] [PubMed]
- Orellana, L.; Yoluk, O.; Carrillo, O.; Orozco, M.; Lindahl, E. Prediction and validation of protein intermediate states from structurally rich ensembles and coarse-grained simulations. Nat. Commun. 2016, 7, 12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poma, A.B.; Li, M.S.; Theodorakis, P.E. Generalization of the elastic network model for the study of large conformational changes in biomolecules. Phys. Chem. Chem. Phys. 2018, 20, 17020–17028. [Google Scholar] [CrossRef] [PubMed]
- Putz, I.; Brock, O. Elastic network model of learned maintained contacts to predict protein motion. PLoS ONE 2017, 12, e0183889. [Google Scholar] [CrossRef] [PubMed]
- Takada, S. Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol. 2012, 22, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Potestio, R.; Pontiggia, F.; Micheletti, C. Coarse-grained description of protein internal dynamics: An optimal strategy for decomposing proteins in rigid subunits. Biophys. J. 2009, 96, 4993–5002. [Google Scholar] [CrossRef] [PubMed]
- Kenzaki, H.; Koga, N.; Hori, N.; Kanada, R.; Li, W.; Okazaki, K.I.; Yao, X.Q.; Takada, S. CafeMol: A coarse-grained biomolecular simulator for simulating proteins at work. J. Chem. Theory Comput. 2011, 7, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.M.; Ahmed, A.; Gohlke, H. NMSim web server: Integrated approach for normal mode-based geometric simulations of biologically relevant conformational transitions in proteins. Nucleic Acids Res. 2012, 40, W310–W316. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Brooks, B.R.; Hummer, G. Protein conformational transitions explored by mixed elastic network models. Proteins Struct. Funct. Bioinform. 2007, 69, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Tekpinar, M.; Zheng, W. Predicting order of conformational changes during protein conformational transitions using an interpolated elastic network model. Proteins Struct. Funct. Bioinform. 2010, 78, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Hollup, S.M.; Salensminde, G.; Reuter, N. WEBnm@: A web application for normal mode analyses of proteins. BMC Bioinform. 2005, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. IMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, E.; Azuara, C.; Koehl, P.; Delarue, M. NOMAD-Ref: Visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006, 34, W52–W56. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Sanejouand, Y.-H. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Y.Y.; Yang, L.W.; Bahar, I. iGNM 2.0: The Gaussian network model database for biomolecular structural dynamics. Nucleic Acids Res. 2016, 44, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.T.; Kloczkowski, A.; Jernigan, R.L. MAVENs: Motion analysis and visualization of elastic networks and structural ensembles. BMC Bioinform. 2011, 12, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Eyal, E.; Lum, G.; Bahar, I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derreumaux, P.; Mousseau, N. Coarse-grained protein molecular dynamics simulations. J. Chem. Phys. 2007, 126, 025101. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.X.; Ingólfsson, H.I.; De Vries, A.H.; Marrink, S.J.; Tieleman, D.P. Ganglioside-lipid and ganglioside-protein interactions revealed by coarse-grained and atomistic molecular dynamics simulations. J. Phys. Chem. B 2017, 121, 3262–3275. [Google Scholar] [CrossRef] [PubMed]
- Gniewek, P.; Kolinski, A.; Jernigan, R.L.; Kloczkowski, A. Elastic network normal modes provide a basis for protein structure refinement. J. Chem. Phys. 2012, 136, 195101. [Google Scholar] [CrossRef] [PubMed]
- Gniewek, P.; Kolinski, A.; Jernigan, R.; Kloczkowski, A. ANM normal modes show the directions for protein structure refinement. Biophys. J. 2012, 102, 25A. [Google Scholar] [CrossRef]
- Schindler, C.E.M.; de Vries, S.J.; Zacharias, M. iATTRACT: Simultaneous global and local interface optimization for protein-protein docking refinement. Proteins Struct. Funct. Bioinform. 2015, 83, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; DiMaio, F.; Baker, D. CASP11 refinement experiments with ROSETTA. Proteins 2016, 84, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Schröder, G.F.; Levitt, M.; Brunger, A.T. Deformable elastic network refinement for low-resolution macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, D70, 2241–2255. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M.; Ben-Avraham, D. Atomic torsional modal analysis for high-resolution proteins. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2015, 91, 032712. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Kanada, R.; Tan, C.; Terakawa, T.; Li, W.; Kenzaki, H. Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Acc. Chem. Res. 2015, 48, 3026–3035. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, V.; Noyes, K.; Feig, M. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins Struct. Funct. Bioinform. 2014, 82, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, V.; Feig, M. Protein structure refinement through structure selection and averaging from molecular dynamics ensembles. J. Chem. Theory Comput. 2013, 9, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Miyashita, O.; Brooks, C.L. Flexible multi-scale fitting of atomic structures into low-resolution electron density maps with elastic network normal mode analysis. J. Mol. Biol. 2004, 337, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Stansfeld, P.J.; Sansom, M.S.P. From coarse grained to atomistic: A serial multiscale approach to membrane protein simulations. J. Chem. Theory Comput. 2011, 7, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Singharoy, A.; Teo, I.; McGreevy, R.; Stone, J.E.; Zhao, J.; Schulten, K. Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps. eLife 2016, 5, e16105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, M.; Mirjalili, V. Protein structure refinement via molecular-dynamics simulations: What works and what does not? Proteins 2016, 84, 282–92. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, A.; Veenhoff, L.M.; Van Der Giessen, E.; Onck, P.R. Probing the disordered domain of the nuclear pore complex through coarse-grained molecular dynamics simulations. Biophys. J. 2014, 107, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Schuyler, A.D.; Jernigan, R.L.; Qasba, P.K.; Ramakrishnan, B.; Chirikjian, G.S. Conformational transition paths are computed by combination and interpolation of normal modes of both strutures. Proteins 2009, 74, 760–776. [Google Scholar] [CrossRef]
- Mahajan, S.; Sanejouand, Y.H. On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys. 2015, 567, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.-W.; Voth, G.A. Coarse-grained free energy functions for studying protein conformational changes: A double-well network model. Biophys. J. 2007, 93, 3860–3871. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tekpinar, M. Protein Dynamics; Analysis of Protein Conformational Transitions Using Elastic Network Model; Humana Press: Totowa, NJ, USA, 2013; pp. 159–172. [Google Scholar]
- Tama, F.; Sanejouand, Y.-H. Conformational change of proteins arising from normal mode calculations. Protein Eng. Des. Sel. 2001, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Frappier, V.; Najmanovich, R.J. A coarse-grained elastic network atom contact model and its use in the simulation of protein dynamics and the prediction of the effect of mutations. PLoS Comput. Biol. 2014, 10, e1003569. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, R.; Sarkar, R.; Kotamarthi, H.C.; Koti, A.S.R. Assessing limitations of elastic network models in describing equilibrium protein flexibility and extensions to predict non-equilibrium unfolding dynamics of proteins. Biophys. J. 2014, 106, 412A. [Google Scholar] [CrossRef]
- Kurkcuoglu, O.; Doruker, P.; Sen, T.Z.; Kloczkowski, A.; Jernigan, R.L. The ribosome structure controls and directs mRNA entry, translocation and exit dynamics. Phys. Biol. 2008, 5, 046005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurkcuoglu, O.; Kurkcuoglu, Z.; Doruker, P.; Jernigan, R.L. Collective dynamics of the ribosomal tunnel revealed by elastic network modeling. Proteins Struct. Funct. Bioinform. 2009, 75, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurkcuoglu, O.; Turgut, O.T.; Cansu, S.; Jernigan, R.L.; Doruker, P. Focused functional dynamics of supramolecules by use of a mixed-resolution elastic network model. Biophys. J. 2009, 97, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kolinski, A.; Skolnick, J. TOUCHSTONE II: A new approach to ab initio protein structure prediction. Biophys. J. 2003, 85, 1145–1164. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Czaplewski, C.; Ołdziej, S.; Scheraga, H.A. Computational techniques for efficient conformational sampling of proteins. Curr. Opin. Struct. Biol. 2008, 18, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaplewski, C.; Karczyńska, A.; Sieradzan, A.K.; Liwo, A. UNRES server for physics-based coarse-grained simulations and prediction of protein structure, dynamics and thermodynamics. Nucleic Acids Res. 2018, 46, W304–W309. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Z.; Lei, T.; Zhou, W.; Chang, S.; Li, D. Importance of protein flexibility on molecular recognition: Modeling binding mechanisms of aminopyrazine inhibitors to Nek2. Phys. Chem. Chem. Phys. 2018, 20, 5591–5605. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Sun, H.; Pan, P.; Zhu, F.; Chang, S.; Xu, L.; Li, Y.; Hou, T. Importance of protein flexibility in molecular recognition: A case study on Type-I1/2 inhibitors of ALK. Phys. Chem. Chem. Phys. 2018, 20, 4851–4863. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, M.; Zotenko, E.; Hildebrandt, A.; Lengauer, T. On the applicability of elastic network normal modes in small-molecule docking. J. Chem. Inf. Model. 2012, 52, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Feig, M. Hybrid All-atom/coarse-grained simulations of proteins by direct coupling of CHARMM and PRIMO force fields. J. Chem. Theory Comput. 2017, 13, 5753–5765. [Google Scholar] [CrossRef] [PubMed]
- Karczyńska, A.S.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Liwo, A.; Czaplewski, C. Prediction of protein structure with the coarse-grained UNRES force field assisted by small X-ray scattering data and knowledge-based information. Proteins Struct. Funct. Bioinform. 2018, 86, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Roux, B. Enhanced sampling of an atomic model with hybrid nonequilibrium molecular dynamics—Monte Carlo simulations guided by a coarse-grained model. J. Chem. Theory Comput. 2015, 11, 3572–3583. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.; Bonvin, A.M. Integrative computational modeling of protein interactions. FEBS J. 2014, 281, 1988–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiga, E.; Degiacomi, M.T.; Dal Peraro, M. New strategies for integrative dynamic modeling of macromolecular assembly. Adv. Protein Chem. Struct. Biol. 2014, 96, 77–111. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Viswanath, S.; Bonomi, M.; Pellarin, R.; Greenberg, C.H.; Saltzberg, D.; Sali, A. Integrative structure modeling with the integrative modeling platform. Protein Sci. 2018, 281, 1988–2003. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kmiecik, S.; Kouza, M.; Badaczewska-Dawid, A.E.; Kloczkowski, A.; Kolinski, A. Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3496. https://doi.org/10.3390/ijms19113496
Kmiecik S, Kouza M, Badaczewska-Dawid AE, Kloczkowski A, Kolinski A. Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. International Journal of Molecular Sciences. 2018; 19(11):3496. https://doi.org/10.3390/ijms19113496
Chicago/Turabian StyleKmiecik, Sebastian, Maksim Kouza, Aleksandra E. Badaczewska-Dawid, Andrzej Kloczkowski, and Andrzej Kolinski. 2018. "Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models" International Journal of Molecular Sciences 19, no. 11: 3496. https://doi.org/10.3390/ijms19113496
APA StyleKmiecik, S., Kouza, M., Badaczewska-Dawid, A. E., Kloczkowski, A., & Kolinski, A. (2018). Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. International Journal of Molecular Sciences, 19(11), 3496. https://doi.org/10.3390/ijms19113496