Role of Overexpressed Transcription Factor FOXO1 in Fatal Cardiovascular Septal Defects in Patau Syndrome: Molecular and Therapeutic Strategies

Abstract
:1. Introduction
Molecular Docking between Candidate Drugs and FOXO1
2. Results
2.1. Cytogenetic Analysis of PS Patient
2.2. Molecular Pathway Analysis
2.3. Genomic Analysis and Protein–Protein Interaction Study
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Cytogenetics Study
4.3. Molecular Pathway and Gene Ontology Analysis
4.4. Identification of Functionally Significant Interacting Proteins of FOXO1
4.5. Molecular Docking and Drug Design
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patau, K.; Smith, D.W.; Therman, E.; Inhorn, S.L.; Wagner, H.P. Multiple congenital anomaly caused by an extra autosome. Lancet (London, England) 1960, 1, 790–793. [Google Scholar] [CrossRef]
- Duarte, A.C.; Menezes, A.I.; Devens, E.S.; Roth, J.M.; Garcias, G.L.; Martino-Roth, M.G. Patau syndrome with a long survival. A case report. Genet. Mol. Res. 2004, 3, 288–292. [Google Scholar] [PubMed]
- Wyllie, J.P.; Wright, M.J.; Burn, J.; Hunter, S. Natural history of trisomy 13. Arch. Dis. Child. 1994, 71, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Pont, S.J.; Robbins, J.M.; Bird, T.M.; Gibson, J.B.; Cleves, M.A.; Tilford, J.M.; Aitken, M.E. Congenital malformations among liveborn infants with trisomies 18 and 13. Am. J. Med. Genet. 2006, 140, 1749–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrelo, A.; Fernandez-Crehuet, P.; Del Prado, E.; Martes, P.; Hernandez-Martin, A.; De Diego, V.; Carapeto, F. Extensive comedonal and cystic acne in Patau syndrome. Pediatr. Dermatol. 2010, 27, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Locock, L.; Crawford, J.; Crawford, J. The parents’ journey: Continuing a pregnancy after a diagnosis of Patau’s syndrome. BMJ 2005, 331, 1186–1189. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.J.; Jacobs, P.A. Trisomy in man. Annu. Rev. Genet. 1984, 18, 69–97. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.F.; Hou, J.W. Variable expressivity in Patau syndrome is not all related to trisomy 13 mosaicism. Am. J. Med. Genet. 2007, 143a, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Sekerli, E.; Vassiliou, G.; Sidiropoulou, V.; Topalidis, A.; Dimopoulou, D.; Voyiatzis, N. Patau syndrome with a long survival (146 months): A clinical report and review of literature. Am. J. Med. Genet. 2006, 140, 92–93. [Google Scholar] [CrossRef] [PubMed]
- Tunca, Y.; Kadandale, J.S.; Pivnick, E.K. Long-term survival in Patau syndrome. Clin. Dysmorphol. 2001, 10, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Redheendran, R.; Neu, R.L.; Bannerman, R.M. Long survival in trisomy-13-syndrome: 21 cases including prolonged survival in two patients 11 and 19 years old. Am. J. Med. Genet. 1981, 8, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Goel, M.; Rathore, R. Trisomy 13 (Patau syndrome). Indian Pediatr. 2000, 37, 1140. [Google Scholar] [PubMed]
- Hassold, T.; Hunt, P. Maternal age and chromosomally abnormal pregnancies: What we know and what we wish we knew. Curr. Opin. Pediatr. 2009, 21, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.C. Trisomy 18 and trisomy 13 syndromes. Manag. Genet. Syndr. 2005. [Google Scholar] [CrossRef]
- Polli, J.B.; Groff Dde, P.; Petry, P.; Mattos, V.F.; Rosa, R.C.; Zen, P.R.; Graziadio, C.; Paskulin, G.A.; Rosa, R.F. Trisomy 13 (Patau syndrome) and congenital heart defects. Am. J. Med. Genet. 2014, 164A, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Yukifumi, M.; Hirohiko, S.; Fukiko, I.; Mariko, M. Trisomy 13 in a 9-year-old girl with left ventricular noncompaction. Pediatr. Cardiol. 2011, 32, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, F. Chromosome 13. Genet. Test. 2000, 4, 85–94. [Google Scholar] [PubMed]
- Auguste, G.; Gurha, P.; Lombardi, R.; Coarfa, C.; Willerson, J.T.; Marian, A.J. Suppression of Activated FOXO Transcription Factors in the Heart Prolongs Survival in a Mouse Model of Laminopathies. Circ. Res. 2018, 122, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Govindsamy, A.; Naidoo, S.; Cerf, M.E. Cardiac Development and Transcription Factors: Insulin Signalling, Insulin Resistance, and Intrauterine Nutritional Programming of Cardiovascular Disease. J. Nutr. Metab. 2018, 2018, 8547976. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Wang, N.; Mao, W.; You, T.; Lu, Y.; Li, X.; Ye, B.; Li, F.; Xu, H. Deletion of FoxO1 leads to shortening of QRS by increasing Na+ channel activity through enhanced expression of both cardiac NaV1. 5 and β3 subunit. J. Mol. Cell. Cardiol. 2014, 74, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ng, F.L.; Chan, K.; Pu, X.; Poston, R.N.; Ren, M.; An, W.; Zhang, R.; Wu, J.; Yan, S.; et al. Coronary-Heart-Disease-Associated Genetic Variant at the COL4A1/COL4A2 Locus Affects COL4A1/COL4A2 Expression, Vascular Cell Survival, Atherosclerotic Plaque Stability and Risk of Myocardial Infarction. PLoS Genet. 2016, 12, e1006127. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Kentrup, D.; Reuter, S.; Mayer, A.B.; Golle, L.; Tiemann, K.; Fobker, M.; Engelbertz, C.; Breithardt, G.; Brand, E.; et al. Soluble Flt-1 links microvascular disease with heart failure in CKD. Basic Res Cardiol. 2015, 110, 30. [Google Scholar] [CrossRef] [PubMed]
- Welten, S.M.; Goossens, E.A.; Quax, P.H.; Nossent, A.Y. The multifactorial nature of microRNAs in vascular remodelling. Cardiovasc. Res. 2016, 110, 6–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzuca, M.Q.; Khalil, R.A. Vascular endothelin receptor type B: Structure, function and dysregulation in vascular disease. Biochem. Pharmacol. 2012, 84, 147–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunbul, M.; Cagman, Z.; Gerin, F.; Ozgen, Z.; Durmus, E.; Seckin, D.; Ahmad, S.; Uras, F.; Agirbasli, M. Growth arrest-specific 6 and cardiometabolic risk factors in patients with psoriasis. Cardiovasc. Ther. 2015, 33, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.; Michaelsson, E.; Linde, C.; Donal, E.; Daubert, J.C.; Gan, L.M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure With Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, N.T.; Shih, Y.P.; Liao, Y.C.; Xue, L.; Lo, S.H. DLC2 modulates angiogenic responses in vascular endothelial cells by regulating cell attachment and migration. Oncogene 2010, 29, 3010–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho Londono, J.E.; Tian, Q.; Hammer, K.; Schroder, L.; Camacho Londono, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Kurtenbach, S.; Kurtenbach, S.; Zoidl, G. Gap junction modulation and its implications for heart function. Front. Physiol. 2014, 5, 82. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Kitayama, K.; Shimoda, Y.; Ogawa, M.; Sone, K.; Yoshida-Araki, K.; Hisatsune, H.; Nishikawa, S.; Nakayama, K.; Nakayama, K.; et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J. Biol. Chem. 2004, 279, 34741–34749. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, Y.A.; Pangas, S.A.; Adams, J.; Zhou, W.; Castrillon, D.H.; Wilhelm, D.; Richards, J.S. FOXO1/3 and PTEN Depletion in Granulosa Cells Promotes Ovarian Granulosa Cell Tumor Development. Mol. Endocrinol. 2015, 29, 1006–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.H.; Collins, S.L.; Sun, I.H.; Tam, A.J.; Patel, C.H.; Arwood, M.L.; Chan-Li, Y.; Powell, J.D.; Horton, M.R. mTORC2 Signaling Selectively Regulates the Generation and Function of Tissue-Resident Peritoneal Macrophages. Cell Rep. 2017, 20, 2439–2454. [Google Scholar] [CrossRef] [PubMed]
- Gomis, R.R.; Alarcon, C.; He, W.; Wang, Q.; Seoane, J.; Lash, A.; Massague, J. A FoxO-Smad synexpression group in human keratinocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12747–12752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herndon, M.K.; Law, N.C.; Donaubauer, E.M.; Kyriss, B.; Hunzicker-Dunn, M. Forkhead box O member FOXO1 regulates the majority of follicle-stimulating hormone responsive genes in ovarian granulosa cells. Mol. Cell Endocrinol. 2016, 434, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lockhart, S.M.; Rathjen, T.; Albadawi, H.; Sorensen, D.; O’Neill, B.T.; Dwivedi, N.; Preil, S.R.; Beck, H.C.; Dunwoodie, S.L.; et al. Insulin Downregulates the Transcriptional Coregulator CITED2, an Inhibitor of Proangiogenic Function in Endothelial Cells. Diabetes 2016, 65, 3680–3690. [Google Scholar] [CrossRef] [PubMed]
- Bround, M.J.; Wambolt, R.; Luciani, D.S.; Kulpa, J.E.; Rodrigues, B.; Brownsey, R.W.; Allard, M.F.; Johnson, J.D. Cardiomyocyte ATP production, metabolic flexibility, and survival require calcium flux through cardiac ryanodine receptors in vivo. J. Biol. Chem. 2013, 288, 18975–18986. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, Y.M.; Mazur, E.C.; Li, X.; Kommagani, R.; Jiang, L.; Chen, R.; Lanz, R.B.; Kovanci, E.; Gibbons, W.E.; DeMayo, F.J. FOXO1 is required for binding of PR on IRF4, novel transcriptional regulator of endometrial stromal decidualization. Mol. Endocrinol. 2015, 29, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa-Hibi, Y.; Kobayashi, Y.; Chen, C.; Motoyama, N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid. Redox Signal. 2005, 7, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Dharaneeswaran, H.; Abid, M.R.; Yuan, L.; Dupuis, D.; Beeler, D.; Spokes, K.C.; Janes, L.; Sciuto, T.; Kang, P.M.; Jaminet, S.S.; et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res. 2014, 115, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Luna-Zurita, L.; Stirnimann, C.U.; Glatt, S.; Kaynak, B.L.; Thomas, S.; Baudin, F.; Samee, M.A.; He, D.; Small, E.M.; Mileikovsky, M.; et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 2016, 164, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.H.; Herrera, R.E.; Coronado-Heinsohn, E.; Yang, M.C.; Ludes-Meyers, J.H.; Seybold-Tilson, K.J.; Nawaz, Z.; Yee, D.; Barr, F.G.; Diab, S.G.; et al. Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor-interacting protein with both coactivator and corepressor functions. J. Biol. Chem. 2001, 276, 27907–27912. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Chakraborty, S.; Paik, J.; Yutzey, K.E.; Evans-Anderson, H.J. FoxO1 is required in endothelial but not myocardial cell lineages during cardiovascular development. Dev. Dyn. 2012, 241, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Huang, Z. FoxO1: A novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget 2017, 8, 10662–10674. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M. Skeletal muscle formation in vertebrates. Curr. Opin. Genet. Dev. 2001, 11, 440–448. [Google Scholar] [CrossRef]
- Gross, D.N.; van den Heuvel, A.P.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiese, K.; Hou, J.; Chong, Z.Z.; Shang, Y.C. A fork in the path: Developing therapeutic inroads with FoxO proteins. Oxid. Med. Cell Longev. 2009, 2, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Puthanveetil, P.; Wan, A.; Rodrigues, B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 2012, 97, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandula, V.; Kosuru, R.; Li, H.; Yan, D.; Zhu, Q.; Lian, Q.; Ge, R.S.; Xia, Z.; Irwin, M.G. Forkhead box transcription factor 1: Role in the pathogenesis of diabetic cardiomyopathy. Cardiovasc. Diabetol. 2016, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Feben, C.; Kromberg, J.; Krause, A. An unusual case of Trisomy 13. S. Afr. J. Child Health 2015, 9, 61–62. [Google Scholar]
- Kajimura, D.; Paone, R.; Mann, J.J.; Karsenty, G. Foxo1 regulates Dbh expression and the activity of the sympathetic nervous system in vivo. Mol. Metab. 2014, 3, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, A.; Battiprolu, P.K.; Ni, Y.G.; Rothermel, B.A.; Hill, J.A. FoxO, autophagy, and cardiac remodeling. J. Cardiovasc. Transl. Res. 2010, 3, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673. [Google Scholar] [CrossRef] [PubMed]
- Mirza, Z.; Beg, M.A. Possible Molecular Interactions of Bexarotene—A Retinoid Drug and Alzheimer’s Abeta Peptide: A Docking Study. Curr. Alzheimer Res. 2017, 14, 327–334. [Google Scholar] [PubMed]
- Tobinick, E.L. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009, 22, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zanella, F.; Dos Santos, N.R.; Link, W. Moving to the Core: Spatiotemporal Analysis of Forkhead Box O (FOXO) and Nuclear Factor-κB (NF-κB) Nuclear Translocation. Traffic 2013, 14, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Obsil, T.; Obsilova, V. Structural basis for DNA recognition by FOXO proteins. Biochim. Biophys. Acta 2011, 1813, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Mutka, S.C.; Yang, W.Q.; Dong, S.D.; Ward, S.L.; Craig, D.A.; Timmermans, P.B.; Murli, S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009, 69, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Kau, T.R.; Schroeder, F.; Ramaswamy, S.; Wojciechowski, C.L.; Zhao, J.J.; Roberts, T.M.; Clardy, J.; Sellers, W.R.; Silver, P.A. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 2003, 4, 463–476. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Huang, H. FOXO1: A potential target for human diseases. Curr. Drug Targets 2011, 12, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.C.; Kau, T.R.; Silver, P.A.; Clardy, J. The psammaplysenes, specific inhibitors of FOXO1a nuclear export. J. Nat. Prod. 2005, 68, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Bain, J.; Elliott, M.; Cohen, P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004, 5, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, W.; Oyarzabal, J.; Serelde, B.G.; Albarran, M.I.; Rabal, O.; Cebriá, A.; Alfonso, P.; Fominaya, J.; Renner, O.; Peregrina, S. Chemical interrogation of FOXO3a nuclear translocation identifies potent and selective inhibitors of phosphoinositide 3-kinases. J. Biol. Chem. 2009, 284, 28392–28400. [Google Scholar] [CrossRef] [PubMed]
- Jaszczyszyn, A.; Gąsiorowski, K.; Świątek, P.; Malinka, W.; Cieślik-Boczula, K.; Petrus, J.; Czarnik-Matusewicz, B. Chemical structure of phenothiazines and their biological activity. Pharmacol. Rep. 2012, 64, 16–23. [Google Scholar] [CrossRef]
- Qi, L.; Ding, Y. Potential antitumor mechanisms of phenothiazine drugs. Sci. China Life Sci. 2013, 56, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-H.; Bai, L.-Y.; Tsai, M.-H.; Chu, P.-C.; Chiu, C.-F.; Chen, M.Y.; Chiu, S.-J.; Chiang, J.-H.; Weng, J.-R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents–A drug repurposing strategy. Sci. Rep. 2016, 6, 27540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Rice, C.M. Repurposing an old drug: A low-cost allergy medication provides new hope for hepatitis C patients. Hepatology 2015, 62, 1911–1913. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.-J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.E.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 10, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Abdelmonem, M.; Zarrin, B.; Asif, N. High Throughput Screening and Molecular Docking of Calmodulin with Antagonists of Trifluoperazine and Phenothiazine Chemical Class. Lett. Drug Des. Discov. 2018, 15, 136–142. [Google Scholar]
- Pan, D.; Yan, Q.; Chen, Y.; McDonald, J.M.; Song, Y. Trifluoperazine Regulation of Calmodulin Binding to Fas: A Computational Study. Proteins 2011, 79, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Yamagishi, H.; Furutani, Y.; Kamisago, M.; Waragai, T.; Oana, S.; Kajino, H.; Matsuura, H.; Mori, K.; Matsuoka, R. The impact of cardiac surgery in patients with trisomy 18 and trisomy 13 in Japan. Am. J. Med. Genet. 2011, 155, 2641–2646. [Google Scholar] [CrossRef] [PubMed]
- Sugayama, S.; Kim, C.; Albano, L.; Utagawa, C.; Bertola, D.; Koiffmann, C. Clinical and genetic study of 20 patients with trisomy 13 (Patau’s syndrome). Pediatria (São Paulo) 1999, 21, 21–29. [Google Scholar]
- Rasmussen, S.A.; Wong, L.-Y.C.; Yang, Q.; May, K.M.; Friedman, J. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics 2003, 111, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Amiel, J.; Claudel, S.; Lyonnet, S.; Corvol, P.; Pinet, F. Functional characterization of three mutations of the endothelin B receptor gene in patients with Hirschsprung’s disease: Evidence for selective loss of Gi coupling. Mol. Med. 2001, 7, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Warburton, D.; Brown, L.Y.; Yu, C.-y.; Roeder, E.R.; Stengel-Rutkowski, S.; Hennekam, R.C.; Muenke, M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat. Genet. 1998, 20, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Paraso, M.; Arkell, R.; Brown, S. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: Alanine tract expansion modulates DNA binding and transactivation. Hum. Mol. Genet. 2004, 14, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Jobanputra, V.; Burke, A.; Kwame, A.Y.; Shanmugham, A.; Shirazi, M.; Brown, S.; Warburton, P.E.; Levy, B.; Warburton, D. Duplication of the ZIC2 gene is not associated with holoprosencephaly. Am. J. Med. Genet. 2012, 158, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Warr, N.; Powles-Glover, N.; Chappell, A.; Robson, J.; Norris, D.; Arkell, R.M. Zic2-associated holoprosencephaly is caused by a transient defect in the organizer region during gastrulation. Hum. Mol. Genet. 2008, 17, 2986–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, B.D.; Lacbawan, F.; Mercier, S.; Clegg, N.J.; Delgado, M.R.; Rosenbaum, K.; Dubourg, C.; David, V.; Olney, A.H.; Wehner, L.-E. Mutations in ZIC2 in human holoprosencephaly: Description of a novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J. Med. Genet. 2010, 47, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barratt, K.S.; Glanville-Jones, H.C.; Arkell, R.M. The Zic2 gene directs the formation and function of node cilia to control cardiac situs. Genesis 2014, 52, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D. The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J. Biol. Chem. 2000, 275, 38949–38952. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, S.; Tokola, H.; Kerkela, R.; Ruskoaho, H. GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 2004, 63, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charron, F.; Paradis, P.; Bronchain, O.; Nemer, G.; Nemer, M. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol. Cell. Biol. 1999, 19, 4355–4365. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Ahuja, S.; Rani, V. Transcription factors in heart: Promising therapeutic targets in cardiac hypertrophy. Curr. Cardiol. Rev. 2011, 7, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Valimaki, M.J.; Tolli, M.A.; Kinnunen, S.M.; Aro, J.; Serpi, R.; Pohjolainen, L.; Talman, V.; Poso, A.; Ruskoaho, H.J. Discovery of Small Molecules Targeting the Synergy of Cardiac Transcription Factors GATA4 and NKX2-5. J. Med. Chem. 2017, 60, 7781–7798. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, S.M.; Tolli, M.; Valimaki, M.J.; Gao, E.; Szabo, Z.; Rysa, J.; Ferreira, M.P.A.; Ohukainen, P.; Serpi, R.; Correia, A.; et al. Cardiac Actions of a Small Molecule Inhibitor Targeting GATA4-NKX2-5 Interaction. Sci. Rep. 2018, 8, 4611. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Ohgi, M.; Koshiba-Takeuchi, K.; Shiratori, H.; Sakaki, I.; Ogura, K.; Saijoh, Y.; Ogura, T. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development 2003, 130, 5953–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boogerd, C.J.; Evans, S.M. TBX5 and NuRD Divide the Heart. Dev. Cell 2016, 36, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Steimle, J.D.; Moskowitz, I.P. TBX5: A Key Regulator of Heart Development. Curr. Top. Dev. Biol. 2017, 122, 195–221. [Google Scholar] [PubMed]
- Stefanovic, S.; Barnett, P.; van Duijvenboden, K.; Weber, D.; Gessler, M.; Christoffels, V.M. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat. Commun. 2014, 5, 3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Hoffmann, A.D.; Burnicka-Turek, O.; Friedland-Little, J.M.; Zhang, K.; Moskowitz, I.P. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wu, X.; Li, Y.; Yang, X.; Hu, J.; Zheng, M.; Tian, J. CITED2 mutation and methylation in children with congenital heart disease. J. Biomed. Sci. 2014, 21, 7. [Google Scholar] [CrossRef] [PubMed]
- Sperling, S.; Grimm, C.H.; Dunkel, I.; Mebus, S.; Sperling, H.P.; Ebner, A.; Galli, R.; Lehrach, H.; Fusch, C.; Berger, F.; et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum. Mutat. 2005, 26, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Chen, R.; Liang, F.; Tsuchiya, H.; Murai, H.; Nakahashi, T.; Iwai, K.; Takahashi, T.; Kanda, T.; Morimoto, S. Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr. Gerontol. Int. 2009, 9, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borradaile, N.M.; Pickering, J.G. NAD(+), sirtuins, and cardiovascular disease. Curr. Pharm. Des. 2009, 15, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Corbi, G.; Bianco, A.; Turchiarelli, V.; Cellurale, M.; Fatica, F.; Daniele, A.; Mazzarella, G.; Ferrara, N. Potential mechanisms linking atherosclerosis and increased cardiovascular risk in COPD: Focus on Sirtuins. Int. J. Mol. Sci. 2013, 14, 12696–12713. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.N.; Wan, M.; Birnbaum, M.J. The role of FOXO in the regulation of metabolism. Curr. Diab. Rep. 2009, 9, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.L. Immune regulation by Foxo transcription factors. Autoimmunity 2007, 40, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Kumar, G.S.; Kadakol, A.; Malek, V.; Bhanudas Gaikwad, A. FoxO1 inhibitors: The future medicine for metabolic disorders? Curr. Diab. Rev. 2016, 12, 223–230. [Google Scholar] [CrossRef]
- Weigelt, J.; Climent, I.; Dahlman-Wright, K.; Wikström, M. Solution structure of the DNA binding domain of the human forkhead transcription factor AFX (FOXO4). Biochemistry 2001, 40, 5861–5869. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Z.; Zhang, M.Q. From worm to human: Bioinformatics approaches to identify FOXO target genes. Mech. Ageing. Dev. 2005, 126, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Nomenclature, I.S.C.o.H.C. ISCN: An International System for Human Cytogenomic Nomenclature (2016); Jean, M.-J., Annet, S., Michael, S., Eds.; Karger: Basel, Switzerland, 2016. [Google Scholar]
- Karim, S.; Merdad, A.; Schulten, H.J.; Jayapal, M.; Dallol, A.; Buhmeida, A.; Al-Thubaity, F.; Mirza, Z.; Gari, M.A.; Chaudhary, A.G.; et al. Low expression of leptin and its association with breast cancer: A transcriptomic study. Oncol. Rep. 2016, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–416. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–815. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Solis, F.J.; Wets, R.J.-B. Minimization by Random Search Techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Cytoband | Associated Disease | Associated Pathways | Paralog |
|---|---|---|---|---|---|
| ATP7B | ATPase Copper Transporting Beta | 13q14.3 | Wilson Disease, Menkes Disease | Cardiac conduction; Ion channel transport; Transmembrane transport of small molecules | ATP7A |
| BRCA2 | Breast cancer 2, early onset | 13q13.1 | Fanconi Anemia, and Breast Cancer | DNA Damage and Role of BRCA1 and BRCA2 in DNA repair | |
| CAB39L | Calcium-binding protein 39-like | 13q14.2 | Acute Monocytic Leukemia | RET signaling and mTOR signaling pathway | CAB39 |
| COL4A1 | Collagen Type IV Alpha 1 Chain | 13q34 | Coronary artery disease | Collagen chain trimerization, Integrin Pathway, ERK Signaling. | COL4A5 |
| DZIP1 | DAZ interacting zinc finger protein 1 | 13q32.1 | Acrodermatitis Enteropathica, Zinc-Deficiency Type | Hedgehog signaling and GPCR signaling. | DZIP1L |
| EDNRB | Endothelin receptor type B | 13q22.3 | Waardenburg Syndrome | Calcium signaling pathway and Prostaglandin Synthesis and Regulation | EDNRA |
| ESD | S-formylglutathione hydrolase | 13q14.2 | Wilson Disease and Leukocoria | Glutathione metabolism | |
| FOXO1 | Forkhead box O 1 | 13q14.11 | Rhabdomyosarcoma 2, Alveolar and Rhabdomyosarcoma | RET signaling; PI3K/AKT activation; Common Cytokine Receptor Gamma-Chain Family Signaling Pathways; AGE/RAGE pathway | FOXO3 |
| FLT1 | Fms-related tyrosine kinase 1 | 13q12.3 | Anal Canal Squamous Cell Carcinoma and Eclampsia | p70S6K Signaling and Focal Adhesion | KDR |
| GAS6 | Growth Arrest Specific 6 | 13q34 | Sticky platelet Syndrome, Acute Maxillary Sinusitis, Mesangial Proliferative Glomerulonephritis | Apoptotic Pathways in Synovial Fibroblasts, GPCR Pathway, ERK Signaling | PROS1 |
| GJB2 | Gap junction protein, beta 2, 26 kDa (connexin 26) | 13q12.11 | Vohwinkel Syndrome and Bart–Pumphrey Syndrome | Development Slit-Robo signaling and Gap junction trafficking. | GJB6. |
| GJB6 | Gap junction protein, beta 6 (connexin 30) | 13q12.11 | Ectodermal Dysplasia 2, Clouston Type and Deafness, Autosomal Dominant 3B | Gap junction trafficking; Vesicle-mediated transport | GJB2 |
| GPC5 | Glypican-5 | 13q31.3 | Simpson–Golabi–Behmel Syndrome and Tetralogy of Fallot | Glycosaminoglycan metabolism | GPC3 |
| HMGB1 | Box 5 Box 1 | 13q12.3 | 13q12.3 Microdeletion Syndrome, Adenosquamous Gallbladder Carcinoma | Activated TLR4 signaling; Cytosolic sensors of pathogen-associated DNA; Innate Immune System | HMGB2 |
| HTR2A | 5-HT2A receptor | 13q14.2 | Schizophrenia; Major Depressive Disorder | Calcium signaling pathway; Signaling by GPCR | HTR2C |
| MIPEP | Mitochondrial intermediate peptidase | 13q12.12 | Combined Oxidative Phosphorylation Deficiency 31 | ||
| PCCA | Propionyl Coenzyme A carboxylase, alpha polypeptide | 13q32.3 | Propionicacidemia and PCCA-Related Propionic Acidemia. | Metabolism and HIV Life Cycle. | MCCC1 |
| RB1 | Retinoblastoma 1 | 13q14.2 | Retinoblastoma and Small-Cell Cancer of the Lung, Somatic. | Arrhythmogenic right ventricular cardiomyopathy (ARVC) and DNA Damage | RBL2 |
| RCBTB1 | RCC1 and BTB domain-containing protein 1 | 13q14.2 | Retinal Dystrophy with Or Without Extraocular Anomalies. | RCBTB2 | |
| RGCC | Regulator of cell cycle RGCC | 13q14.11 | Renal Fibrosis and Retinal Cancer | TP53 Regulates Transcription of Cell Cycle Genes | |
| RNR1 | Encoding RNA, ribosomal 45S cluster 1 | 13p12 | Idiopathic Bilateral Vestibulopathy and Congenital Cytomegalovirus | Viral mRNA Translation | |
| SLITRK6 | SLIT and NTRK-like protein 6 | 13q31.1 | Deafness and Yopia and Autosomal Recessive Non-Syndromic Sensorineural Deafness | SLITRK5 | |
| SOX21 | Transcription factor SOX-21 | 13q32.1 | Mesodermal Commitment Pathway and ERK Signaling. | Mesodermal Commitment Pathway; ERK Signaling | SOX14 |
| STARD13 | StAR-Related Lipid Transfer Domain Containing 13 | 13q13 | Hepatocellular Carcinoma, Arteriovenous Malformations of the Brain, Fibrosarcoma of Bone | p75 NTR receptor-mediated signaling, Signaling by GPCR, Signaling by Rho GTPases | STARD8 |
| TPT1 | Translationally controlled tumor protein (TCTP) | 13q14.13 | Urticaria and Asthma | DNA Damage and Cytoskeletal Signaling | |
| TRPC4 | Transient Receptor Potential Cation Channel Subfamily C Member 4 | 13q13.3 | Photosensitive Epilepsy | Developmental Biology, Ion channel transport, Netrin-1 signaling | TRPC5 |
| TSC22D1 | TSC22 domain family protein 1 | 13q14.11 | Salivary Gland Cancer and Brain Sarcoma | Development TGF-beta receptor signaling and Ectoderm Differentiation | TSC22D2 |
| TUBA3C | Tubulin Alpha 3C | 13q12.11 | Clouston Syndrome, nonsyndromic Deafness, Kabuki Syndrome 1 | Development Slit-Robo signaling, Cooperation of Prefoldin and TriC/CCT in actin and tubulin folding | TUNA3D |
| XPO4 | Exportin-4 | 13q12.11 | Conjunctival Degeneration and Pinguecula | eIF5A regulation in response to inhibition of the nuclear export system and Ran Pathway | |
| ZIC2 | Zic Family Member 2 | 13q32.3 | Holoprosencephaly 5 and Zic2-Related Holoprosencephaly | Mesodermal Commitment Pathway | ZIC1 |
| ZMYM2 | Zinc finger MYM-type protein 2 | 13q12.11 | Lymphoblastic Lymphoma and 8P11 Myeloproliferative Syndrome | HIV Life Cycle and FGFR1 mutant receptor activation | ZMYM3 |
| Canonical Pathways | −log (p Value) | Ratio | Molecules |
|---|---|---|---|
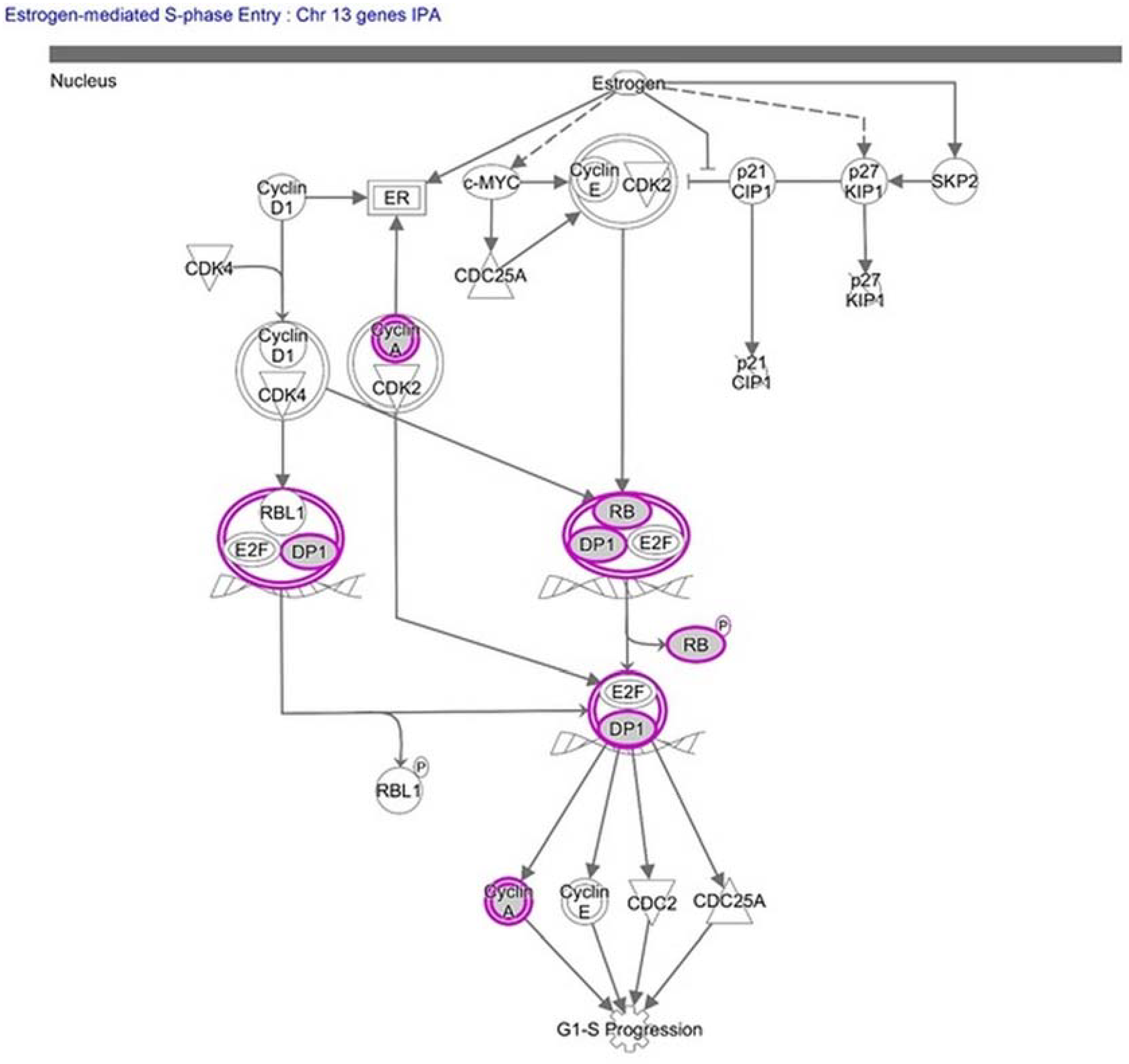
| Estrogen-mediated S-phase Entry | 2.06 | 0.115 | RB1, CCNA1, TFDP1 |
| Cancer Signaling | 1.69 | 0.052 | RB1, FOXO1, TFDP1, KL, IRS2, CDK8, SMAD9, TFDP1, ARHGEF7 |
| Extrinsic Prothrombin Activation Pathway | 1.56 | 0.125 | F10, F7 |
| Role of p14/p19ARF in Tumor Suppression | 1.5 | 0.071 | RB1, KL, IRS2 |
| Gap Junction Signaling | 1.41 | 0.036 | GJB6, KL, GJA3, TUBA3C/TUBA3D, IRS2, GJB2, HTR2A |
| Docosahexaenoic Acid (DHA) Signaling | 1.27 | 0.057 | FOXO1, KL, IRS2 |
| Aldosterone Signaling in Epithelial Cells | 1.24 | 0.035 | SACS, KL, HSPH1, DNAJC3, IRS2, DNAJC15 |
| FGF Signaling | 1.2 | 0.044 | KL, FGF9, FGF14, IRS2 |
| GP6 Signaling Pathway | 1.18 | 0.038 | COL4A1, KL, IRS2, COL4A2, KLF12 |
| Adipogenesis pathway | 1.17 | 0.037 | RB1, SAP18, SMAD9, FOXO1, KLF5 |
| VEGF Signaling | 1.08 | 0.040 | FOXO1, FLT1, KL, IRS2 |
| Cell Cycle: G1/S Checkpoint Regulation | 1.04 | 0.046 | RB1, FOXO1, TFDP1 |
| ErbB2-ErbB3 Signaling | 0.994 | 0.044 | FOXO1, KL, IRS2 |
| Nitric Oxide Signaling in the Cardiovascular System | 0.988 | 0.037 | FLT1, KL, SLC7A1, IRS2 |
| Coagulation System | 0.948 | 0.057 | F10, F7 |
| Angiopoietin Signaling | 0.875 | 0.039 | FOXO1, KL, IRS2 |
| Role of NANOG in Mammalian Embryonic Stem Cell Pluripotency | 0.866 | 0.0333 | SMAD9, KL, CDX2, IRS2 |
| IL-3 Signaling | 0.805 | 0.036 | FOXO1, KL, IRS2 |
| Actin Cytoskeleton Signaling | 0.801 | 0.027 | KL, FGF9, DIAPH3, ARHGEF7, FGF14, IRS2 |
| 14-3-3-mediated Signaling | 0.778 | 0.030 | FOXO1, KL, TUBA3C/TUBA3D, IRS2 |
| IL-7 Signaling Pathway | 0.774 | 0.034 | FOXO1, KL, IRS2 |
| HMGB1 Signaling | 0.77 | 0.030 | HMGB1, KL, IL17D, IRS2 |
| NF-κB Signaling | 0.769 | 0.028 | TNFSF11, FLT1, KL, IRS2, TNFSF13B |
| Gene Symbol | Gene Name | Cytoband | Associated Disease | Associated Pathways | Paralog |
|---|---|---|---|---|---|
| NODAL | Nodal Growth Differentiation Factor | 10q22 | Visceral Heterotaxy 5 (HTX5) and Nodal-Related Visceral Heterotaxy | Mesodermal Commitment Pathway and Signaling pathways regulating pluripotency of stem cells | GDF3 |
| FPR1 | Formyl Peptide Receptor 1 | 19q13.41 | Susceptibility to Localized Juvenile Periodontitis and Periodontitis 1, Juvenile | Signaling by GPCR and Peptide ligand-binding receptors | FPR2 |
| AFP | Alpha Fetoprotein | 4q13.3 | Alpha-Fetoprotein Deficiency and Hereditary Persistence of Alpha-Fetoprotein | Glucocorticoid receptor regulatory network and Embryonic and Induced Pluripotent Stem Cell Differentiation Pathways and Lineage-specific Markers | ALB |
| AGO2 | Argonaute RISC Catalytic Component 2 | 8q24.3 | Chromosome 18P Deletion Syndrome and Gum Cancer | RET signaling and Translational Control. | AGO1 |
| UROD | Uroporphyrinogen Decarboxylase | 1p34.1 | Porphyria Cutanea Tarda and Urod-Related Porphyrias | Metabolism and Porphyrin and chlorophyll metabolism | |
| GATA4 | GATA Binding Protein 4 | 8p23.1 | Testicular Anomalies with or without Congenital Heart Disease and Atrial Septal Defect 2 | Response to elevated platelet cytosolic Ca2+ and Human Embryonic Stem Cell Pluripotency | GATA6 |
| GATA6 | GATA Binding Protein 6 | 18q11.2 | Pancreatic Agenesis and Congenital Heart Defects and Atrioventricular Septal Defect 5 | Mesodermal Commitment Pathway and Response to elevated platelet cytosolic Ca2+ | GATA4 |
| GJA1 | Gap Junction Protein Alpha 1 | 6q22.31 | Oculodentodigital Dysplasia and Syndactyly, Type Iii | Development Slit-Robo signaling and Arrhythmogenic right ventricular cardiomyopathy | GJA3 |
| JAG1 | Jagged 1 | 20p12.2 | Alagille Syndrome 1 and Tetralogy of Fallot | Signaling by NOTCH1 and NOTCH2 Activation and Transmission of Signal to the Nucleus | JAG2 |
| CITED2 | Cbp/P300 Interacting Transactivator with Glu/Asp Rich Carboxy-Terminal Domain2 | 6q24.1 | Atrial Septal Defect 8 and Ventricular Septal Defect 2 | Cellular Senescence (REACTOME) and Transcriptional regulation by the AP-2 (TFAP2) family of transcription factors | CITED1 |
| RYR2 | Ryanodine Receptor 2 | 1q43 | Ventricular Tachycardia, Catecholaminergic Polymorphic, 1 and Arrhythmogenic Right Ventricular Dysplasia 2 | Calcium signaling pathway and Arrhythmogenic right ventricular cardiomyopathy | RYR3 |
| NKX2-5 | NK2 Homeobox 5 | 5q35.1 | Atrial Septal Defect 7, With or Without Av Conduction Defects and Tetralogy of Fallot | Human Embryonic Stem Cell Pluripotency and NFAT and Cardiac Hypertrophy | NKX2-3 |
| RARA | Retinoic Acid Receptor Alpha | 17q21.2 | Leukemia, Acute Promyelocytic, Somatic and Myeloid Leukemia | Nuclear Receptors in Lipid Metabolism and Toxicity and Activated PKN1 stimulates transcription of AR (androgen receptor) regulated genes KLK2 and KLK3. | RARB |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 | 10q11.21 | HIV-1 and AIDS Dementia Complex | p70S6K Signaling and Akt Signaling | |
| SIRT1 | Sirtuin 1 | 10q21.3 | Xeroderma Pigmentosum, Group D and Ovarian Endodermal Sinus Tumor | Longevity regulating pathway and E2F transcription factor network | SIRT4 |
| TBX5 | T-Box 5 | 12q24.21 | Holt–Oram Syndrome and Aortic Valve Disease 2 | Human Embryonic Stem Cell Pluripotency and Cardiac conduction. | TBX4 |
| AKT1 | AKT Serine/Threonine Kinase 1 | 14q32.33 | Cowden Syndrome 6 and Proteus Syndrome, Somatic | Transcription Androgen Receptor nuclear signaling and E-cadherin signaling in keratinocytes | AKT3 |
| CDKN2A | Cyclin Dependent Kinase Inhibitor 2A | 9p21.3 | Pancreatic Cancer/Melanoma Syndrome and Melanoma and Neural System Tumor Syndrome | DNA Damage and Bladder cancer | CDKN2B |
| PCK1 | Phosphoenolpyruvate Carboxykinase 1 | 20q13.31 | Pepck 1 Deficiency and Phosphoenolpyruvate Carboxykinase-1, Cytosolic, Deficiency | Abacavir transport and metabolism and Citrate cycle (TCA cycle) | PCK2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuzenadah, A.; Alsaedi, S.; Karim, S.; Al-Qahtani, M. Role of Overexpressed Transcription Factor FOXO1 in Fatal Cardiovascular Septal Defects in Patau Syndrome: Molecular and Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 3547. https://doi.org/10.3390/ijms19113547
Abuzenadah A, Alsaedi S, Karim S, Al-Qahtani M. Role of Overexpressed Transcription Factor FOXO1 in Fatal Cardiovascular Septal Defects in Patau Syndrome: Molecular and Therapeutic Strategies. International Journal of Molecular Sciences. 2018; 19(11):3547. https://doi.org/10.3390/ijms19113547
Chicago/Turabian StyleAbuzenadah, Adel, Saad Alsaedi, Sajjad Karim, and Mohammed Al-Qahtani. 2018. "Role of Overexpressed Transcription Factor FOXO1 in Fatal Cardiovascular Septal Defects in Patau Syndrome: Molecular and Therapeutic Strategies" International Journal of Molecular Sciences 19, no. 11: 3547. https://doi.org/10.3390/ijms19113547
APA StyleAbuzenadah, A., Alsaedi, S., Karim, S., & Al-Qahtani, M. (2018). Role of Overexpressed Transcription Factor FOXO1 in Fatal Cardiovascular Septal Defects in Patau Syndrome: Molecular and Therapeutic Strategies. International Journal of Molecular Sciences, 19(11), 3547. https://doi.org/10.3390/ijms19113547