HGF/MET and the Immune System: Relevance for Cancer Immunotherapy

 ,
, 

Abstract
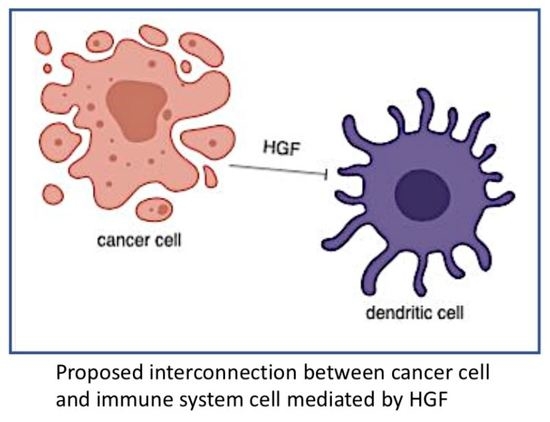
:
{kind=link}
{kind=link}
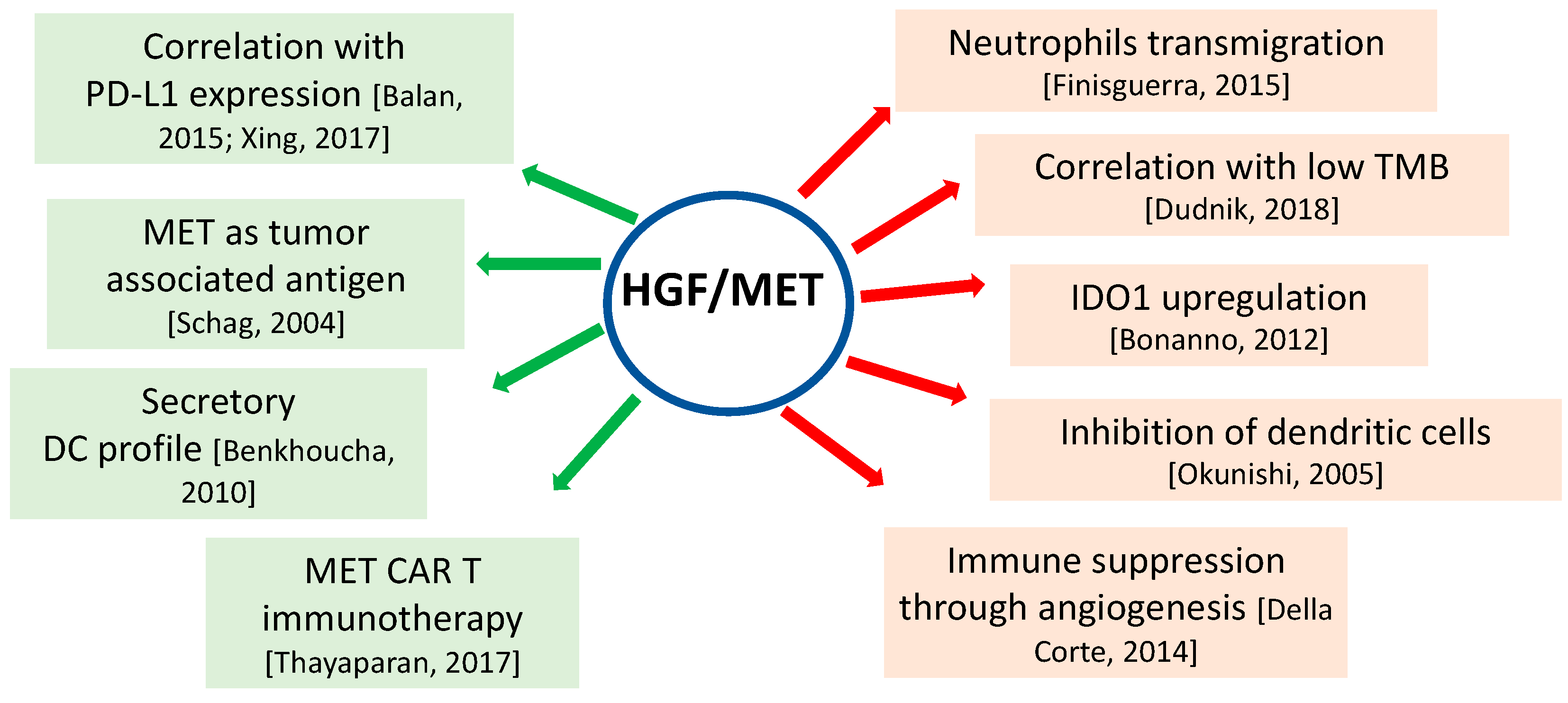{kind=link}
1. Introduction
2. Relevant Immune Pathways for Cancer Treatment
3. HGF/MET and Immune System in Cancer
4. Implications of HGF/MET in Anti-Cancer Immunotherap
5. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aparicio, I.M.; Garcia-Marin, L.J.; Andreolotti, A.G.; Bodega, G.; Jensen, R.T.; Bragado, M.J. Hepatocyte growth factor activates several transduction pathways in rat pancreatic acini. Biochim. Biophys. Acta 2003, 1643, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Nakayama, M.; Suzuki, Y.; Sakai, K.; Nakamura, T.; Sakai, Y.; Matsumoto, K. Suppression of acute hepatic injury by a synthetic prostacyclin agonist through hepatocyte growth factor expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G420–G429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Traut-wein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphoring receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, L.J.; El-Hefnawy, T.; Queiroz de Oliveira, P.E.; Raja, S.; Finkelstein, S.; Gooding, W.; Luketich, J.D.; Godfrey, T.E.; Hughes, S.J. The HGF receptor c-Met is overexpressed in esophageal adenocarcinoma. Neoplasia 2005, 7, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.L.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Choi, Y.L.; Sung, C.O.; An, J.; Seo, J.; Ahn, M.J.; Ahn, J.S.; Park, K.; Shin, Y.K.; Erkin, O.C.; et al. High MET copy number and MET overexpression: Poor outcome in non-small cell lung cancer patients. Histol. Histopathol. 2012, 27, 197–207. [Google Scholar] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Napolitano, S.; Della Corte, C.M.; Martini, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open 2016, 1, e000088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, J.R.; Tsao, M.S. c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3, S21–S35. [Google Scholar] [CrossRef] [PubMed]
- Daveau, M.; Scotte, M.; François, A.; Coulouarn, C.; Ros, G.; Tallet, Y.; Hiron, M.; Hellot, M.F.; Salier, J.P. Hepatocyte growth factor; transforming growth factor alpha; and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol. Carcinog. 2003, 36, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Prechtel, D.; Resau, J.H.; Gauger, K.; Welk, A.; Lindemann, K.; Salanti, G.; Richter, T.; Knudsen, B.; vande Woude, G.F.; et al. c-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome inde pendent of Her2/neu. Int. J. Cancer 2005, 113, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Bilchik, A.; Saha, S.; Turner, R.; Wiese, D.; Tanaka, M.; Kuo, C.; Wang, H.J.; Hoon, D.S. c-MET expression level in primary colon cancer: A predictor of tumor invasion and lymphnode metastases. Clin. Cancer Res. 2003, 9, 1480–1488. [Google Scholar] [PubMed]
- Della Corte, C.M.; Fasano, M.; Papaccio, F.; Ciardiello, F.; Morgillo, F. Role of HGF-MET signaling in primary and acquired resistance to targeted therapies in cancer. Biomedicines 2014, 2, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Amendola, G.; Della Corte, C.M.; Giacomelli, C.; Botta, L.; Di Maro, S.; Messere, A.; Ciaramella, V.; Taliani, S.; Marinelli, L.; et al. Dual MET and SMO negative modulators overcome resistance to EGFR inhibitors in human nonsmall cell lung cancer. J. Med. Chem. 2017, 60, 7447–7458. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Choudhury, A.; Lladser, A.; Kiessling, R.; Johansson, C.C. Regulatory T cells in cancer. Adv. Cancer Res. 2010, 107, 57–117. [Google Scholar] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Carrizosa, D.R.; Gold, K.A. New strategies in immunotherapy for non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 553–559. [Google Scholar] [PubMed]
- Brahmer, J.R.; Rizvi, N.A.; Lutzky, J.; Khleif, S.; Blake-Haskins, A.; Li, X.; Robbins, P.B.; Vasselli, J.; Ibrahim, R.A.; Antonia, S.J. Clinical activity and biomarkers of MEDI4736, an anti-PD-L1 antibody, in patients with NSCLC. J. Clin. Oncol. 2014, 32, 8021. [Google Scholar] [CrossRef]
- Croft, M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003, 14, 265–273. [Google Scholar] [CrossRef]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Kaneda, Y.; Tsutsui, H.; Nakanishi, K.; Sawa, Y.; Morishita, R.; Matsumoto, K.; Nakamura, T.; Takahashi, H.; Okamoto, E.; et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat. Med. 1999, 5, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Dolcet, X.; Hilton, M.; Tolcos, M.; Davies, A.M. HGF promotes survival and growth of maturing sympathetic neurons by PI-3 kinase- and MAP kinase-dependent mechanisms. Mol. Cell Neurosci. 2004, 27, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, K.; Ito, K.; Tsukioka, K.; Wada, Y.; Makiuchi, A.; Sakaguchi, M.; Akashima, T.; Fujimori, M.; Sawa, Y.; Morishita, R.; et al. Suppression of acute and chronic rejection by hepatocyte growth factor in a murine model of cardiac transplantation: Induction of tolerance and prevention of cardiac allograft vasculopathy. Circulation 2004, 110, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Benkhoucha, M.; Santiago-Raber, M.L.; Schneiter, G.; Chofflon, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6424–6429. [Google Scholar] [CrossRef] [PubMed]
- Okunishi, K.; Dohi, M.; Nakagome, K.; Tanaka, R.; Mizuno, S.; Matsumoto, K.; Miyazaki, J.; Nakamura, T.; Yamamoto, K. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J. Immunol. 2005, 175, 4745–4753. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Birchmeier, C.; Zenke, M.; Hieronymus, T. The HGF receptor/Met tyrosine kinase is a key regulator of dendritic cell migration in skin immunity. J. Immunol. 2012, 189, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- van der Voort, R.; Taher, T.E.; Keehnen, R.M.; Smit, L.; Groenink, M.; Pals, S.T. Paracrine regulation of germinal center B cell adhesion through the c-Met-hepatocyte growth factor/scatter factor pathway. J. Exp. Med. 1997, 185, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Harvatht, L.; Bottaro, D.P.; Interranteo, R.; Catalanot, G.; Tanaka, Y.; Strain, A.; Hubscheri, S.G.; Shaw, S. Hepatocyte growth factor and macrophage inflammatory protein 1 beta: Structurally distinct cytokines that induce rapid cytoskeletal changes and subset-preferential migration in T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 7144–7148. [Google Scholar] [CrossRef] [PubMed]
- Kurz, S.M.; Diebold, S.S.; Hieronymus, T.; Gust, T.C.; Bartunek, P.; Sachs, M.; Birchmeier, W.; Zenke, M. The impact of c-Met/scatter factor receptor on dendritic cell migration. Eur. J. Immunol. 2002, 32, 1832–1838. [Google Scholar] [CrossRef]
- Mizuno, S.; Kurosawa, T.; Matsumoto, K.; Mizuno-Horikawa, Y.; Okamoto, M.; Nakamura, T. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J. Clin. Investig. 1998, 101, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, A.P.; Shah, R.; Hochwald, G.D.; Liggitt, H.D.; Palladino, M.A.; Thorbecke, G.J. Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 2918–2921. [Google Scholar] [CrossRef] [PubMed]
- Futamatsu, H.; Suzuki, J.; Mizuno, S.; Koga, N.; Adachi, S.; Kosuge, H.; Maejima, Y.; Hirao, K.; Nakamura, T.; Isobe, M. Hepatocyte growth factor ameliorates the progression of experimental autoimmune myocarditis: A potential role for induction of T helper 2 cytokines. Circ. Res. 2005, 96, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Matsumoto, K.; Li, M.Y.; Nakamura, T. HGF reduces advancing lung fibrosis in mice: A potential role for MMP-dependent myofibroblast apoptosis. FASEB J. 2015, 19, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Ilangumaran, S.; Villalobos-Hernandez, A.; Bobbala, D.; Ramanathan, S. The hepatocyte growth factor (HGF)-MET receptor tyrosine kinase signaling pathway: Diverse roles in modulating immune cell functions. Cytokine 2016, 82, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Canadas, I.; Rojo, F.; Taus, A.; Arpi, O.; Arumi-Uria, M.; Pijuan, L.; Menendez, S.; Zazo, S.; Domine, M.; Salido, M.; et al. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin. Cancer Res. 2014, 20, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Schag, K.; Schmidt, S.M.; Muller, M.R.; Weinschenk, T.; Appel, S.; Weck, M.M.; Grunebach, F.; Stevanovic, S.; Rammensee, H.G.; Brossart, P. Identification of C-met onco-gene as a broadly expressed tumor-associated antigen recognized by cytotoxic T-lymphocytes. Clin. Cancer Res. 2004, 10, 3658–3666. [Google Scholar] [CrossRef] [PubMed]
- Galimi, F.; Cottone, E.; Vigna, E.; Arena, N.; Boccaccio, C.; Giordano, S.; Naldini, L.; Comoglio, P.M. Hepatocyte growth factor is a regulator of monocyte-macrophage function. J. Immunol. 2001, 166, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Hilkens, C.M.; Kalinski, P.; de Boer, M.; Kapsenberg, M.L. Human dendritic cells require exogenous interleukin-12-inducing factors to direct the development of naive T-helper cells toward the Th1 phenotype. Blood 1997, 90, 1920–1926. [Google Scholar] [PubMed]
- Rutella, S.; Bonanno, G.; Procoli, A.; Mariotti, A.; de Ritis, D.G.; Curti, A.; Danese, S.; Pessina, G.; Pandolfi, S.; Natoni, F.; et al. Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features. Blood 2006, 108, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, M.; Teran, E.M.Y.; Waaga-Gasser, A.M.; Gasser, M.; Choueiri, T.K.; Freeman, G.; Pal, S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J. Biol. Chem. 2015, 290, 8110–8120. [Google Scholar] [CrossRef] [PubMed]
- Singhal, E.; Sen, P. Hepatocyte growth factor-induced c-Src-phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin pathway inhibits dendritic cell activation by blocking IκB kinase activity. Int. J. Biochem. Cell Biol. 2011, 43, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Singhal, E.; Kumar, P.; Sen, P. A novel role for Bruton’s tyrosine kinase in hepatocyte growth factor-mediated immunoregulation of dendritic cells. J. Biol. Chem. 2011, 286, 32054–32063. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, G.; Mariotti, A.; Procoli, A.; Folgiero, V.; Natale, D.; De Rosa, L.; Majolino, I.; Novarese, L.; Rocci, A.; Gambella, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity correlates with immune system abnormalities in multiple myeloma. J. Transl. Med. 2012, 10, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanackovic, D.; Luetkens, T.; Kroger, N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia 2014, 28, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Novello, S.; Schiller, J.H.; Hirsh, V.; Sequist, L.V.; Soria, J.-C.; von Pawel, J.; Schwartz, B.; Von Roemeling, R.; Sandler, A.B. Rationale and design of MARQUEE: A phase III randomized; double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic; nonsquamous; non-small-cell lung cancer. Clin. Lung Cancer 2012, 13, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Govindan, R.; Zvirbule, Z.; Braiteh, F.; Rittmeyer, A.; Belda-Iniesta, C.; Isla, D.; Cosgriff, T.; Boyer, M.; Ueda, M.; et al. Efficacy and safety results from a phase II, placebo-controlled study of onartuzumab plus first-line platinum-doublet chemotherapy for advanced squamous cell non-small-cell lung cancer. Clin. Lung Cancer 2017, 18, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Campone, M.; Yardley, D.A.; Romieu, G.; Valero, V.; Isakoff, S.J.; Koeppen, H.; Wilson, T.R.; Xiao, Y.; Shames, D.S.; et al. Randomized phase II, placebo-controlled trial of onartuzumab and/or bevacizumab in combination with weekly paclitaxel in patients with metastatic triple-negative breast cancer. Ann. Oncol. 2015, 26, 1904–1910. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.T.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I.; et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1467–1482. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Stenehjem, D.D.; Tran, D.; Nkrumah, M.A.; Gupta, S. PD1/PDL1 inhibitors for the treatment of advanced urothelial bladder cancer. OncoTargets Ther. 2018, 11, 5973–5989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Medeiros, L.J.; Young, K.H. Cancer immunotherapy in diffuse large B-cell lymphoma. Front. Oncol. 2018, 8, 351. [Google Scholar] [CrossRef] [PubMed]
- de Marinis, F.; Ciardiello, F.; Baas, P.; Crinò, L.; Giaccone, G.; Grossi, F.; Hellmann, M.D.; Mok, T.S.K.; Lena, H.; Paz-Ares, L.; et al. 30 Immunotherapy in advanced NSCLC-from the ‘tsunami’ of therapeutic knowledge to a clinical practice algorithm: Results from an international expert panel meeting of the Italian Association of Thoracic Oncology (AIOT). ESMO Open 2018, 3, e000298. [Google Scholar] [CrossRef] [PubMed]
- Vanella, V.; Festino, L.; Strudel, M.; Simeone, E.; Grimaldi, A.M.; Ascierto, P.A. PD-L1 inhibitors in the pipeline: Promise and progress. Oncoimmunology 2017, 7, e1365209. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Ou, S.-H.; Clark, J.; Camidge, D.R.; Socinski, M.; Weiss, J.; Solomon, B.; Riely, G.; Heist, R.; Shapiro, G.; et al. MA16.09 antitumor activity and safety of crizotinib in patients with advanced MET exon 14-altered non-small cell lung cancer. J. Thorac Oncol. 2017, 12, S438–S439. [Google Scholar] [CrossRef]
- Sabari, J.K.; Montecalvo, J.; Chen, R.; Dienstag, J.A.; Mrad, C.; Bergagnini, I.; Lai, W.-C.V.; Arbour, K.C.; Shu, C.A.; Hellmann, M.D.; et al. PD-L1 expression and response to immunotherapy in patients with MET exon 14-altered non-small cell lung cancers (NSCLC). J. Clin. Oncol. 2017, 35, 8512. [Google Scholar] [CrossRef]
- Dudnik, E.; Peled, N.; Nechushtan, H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF mutant lung cancer: Programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J. Thorac. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Guo, J.; Wen, X.; Ding, G.; Li, B.; Dong, B.; Feng, Q.; Li, S.; Zhang, J.; Cheng, X.; et al. Analysis of PD1, PDL1, PDL2 expression and T cells infiltration in 1014 gastric cancer patients. Oncoimmunology 2017, 7, e1356144. [Google Scholar] [CrossRef] [PubMed]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity 2017, 47, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kumai, T.; Matsuda, Y.; Ohkuri, T.; Oikawa, K.; Ishibashi, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology 2015, 4, e976077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.J.; Wu, Y.; Hou, W.H.; Wang, Y.; Yuan, Q.Y.; Wang, H.J.; Yu, M. A novel bispecific c-MET/PD-1 antibody with therapeutic potential in solid cancer. Oncotarget 2017, 8, 29067–29079. [Google Scholar] [CrossRef] [PubMed]
- Thayaparan, T.; Petrovic, R.M.; Achkova, D.Y.; Zabinski, T.; Davies, D.M.; Klampatsa, A.; Parente-Pereira, A.C.; Whilding, L.M.; van der Stegen, S.J.; Woodman, N.; et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology 2017, 6, e1363137. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.P.; Kros, J.M.; Li, J. Approved CAR T cell therapies: Ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 2018, 23, 1175–11828. [Google Scholar] [CrossRef] [PubMed]
- Halme, D.G.; Kessler, D.A. FDA regulation of stem-cell-based therapies. N. Engl. J. Med. 2006, 355, 1730–1735. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D., Jr.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudjafari, Z.; Hawks, K.G.; Hsieh, A.A.; Plesca, D.; Gatwood, K.S.; Culos, K.A. American Society for Blood and Marrow Transplantation Pharmacy Special Interest Group Survey on Chimeric Antigen Receptor T Cell Therapy Administrative, Logistic, and Toxicity Management Practices in the United States. Biol. Blood Marrow Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, J.; Hu, Q.; Hochu, G.M.; Xin, H.; Wang, C.; Gu, Z. Synergistic transcutaneous immunotherapy enhances antitumor immune responses through delivery of checkpoint inhibitors. ACS Nano 2016, 10, 8956–8963. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, Y.; Hochu, G.M.; Sadeghifar, H.; Gu, Z. Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Lett. 2016, 16, 2334–2340. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papaccio, F.; Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. https://doi.org/10.3390/ijms19113595
Papaccio F, Della Corte CM, Viscardi G, Di Liello R, Esposito G, Sparano F, Ciardiello F, Morgillo F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. International Journal of Molecular Sciences. 2018; 19(11):3595. https://doi.org/10.3390/ijms19113595
Chicago/Turabian StylePapaccio, Federica, Carminia Maria Della Corte, Giuseppe Viscardi, Raimondo Di Liello, Giovanna Esposito, Francesca Sparano, Fortunato Ciardiello, and Floriana Morgillo. 2018. "HGF/MET and the Immune System: Relevance for Cancer Immunotherapy" International Journal of Molecular Sciences 19, no. 11: 3595. https://doi.org/10.3390/ijms19113595
APA StylePapaccio, F., Della Corte, C. M., Viscardi, G., Di Liello, R., Esposito, G., Sparano, F., Ciardiello, F., & Morgillo, F. (2018). HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. International Journal of Molecular Sciences, 19(11), 3595. https://doi.org/10.3390/ijms19113595