3. Discussion
It has been reported that activated human T cells cultured in RWVs present alterations in expression of genes involved in several cellular processes such as signal transduction, DNA repair, apoptosis, immune and inflammatory responses, and metabolic pathways [
55]. Decreased expression of genes involved in mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER) as well as downregulation of p53 were observed in lymphocytes grown in simulated microgravity [
16]. Our results are apparently contradictory to these findings, but it should be noted that the above-mentioned studies were conducted in proliferating lymphocytes. However, most of the time immune cells are in a quiescent, nonproliferating state. Nonetheless, our previous knowledge on DNA repair mechanisms has been gained, to a large extent, from studies on actively growing cells and little is known about how cells in the quiescent state repair DNA. Recent findings demonstrated attenuation of DNA repair in quiescent hematopoietic stem cells leading to accumulation of DNA damage, which is repaired upon entry into cell cycle [
56]. Furthermore, circulating human B lymphocytes are deficient in nucleotide excision repair (NER) [
57], and the ATR-p53 pathway is suppressed in noncycling lymphocytes via ATR downregulation [
58]. Mitogen stimulation might also influence the DNA damage response. For instance, phosphorylation of the histone H2AX and ATM activation were strongly amplified during mitogenic stimulation of lymphocytes [
59]. Interestingly, microgravity forces growing cells into a condition of metabolic quiescence and strongly affects energy metabolism and DNA repair [
17]. Thus, it could be argued that gravity-associated differences in DNA repair pathways can be attributed to the propensity of microgravity to inhibit cell proliferation resulting in an increase in quiescent cells. Since in our experiments we did not (intentionally) stimulate PBMCs to grow, this hypothesis would explain our lack of results supporting gravity-associated changes in gene expression. However, further studies are necessary in order to clarify this observation.
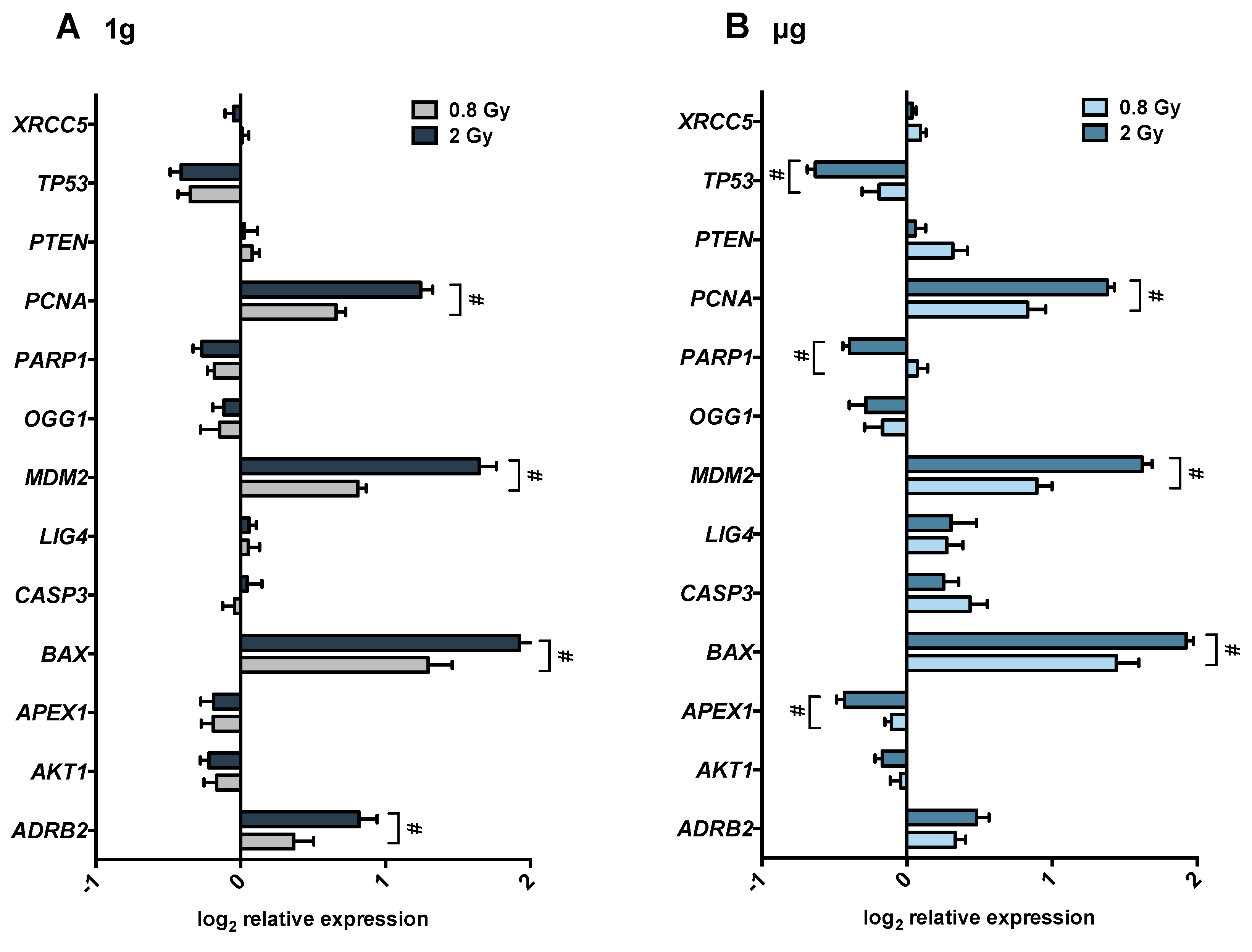
We found a significant synergy between isoproterenol, microgravity and radiation in their combined effect on
ADBR2 expression. This finding suggests a novel potential player in the regulation of radiation-induced DNA damage response, namely the involvement of β2-adrenergic receptor activation. In particular, we found an increase in ADRB2 mRNA levels after isoproterenol treatment in 1
g, which was reduced in µg. Upregulation of
ADRB2 gene expression can lead to a higher receptor density and consequently to a higher adrenergic sensitivity. Indeed, acute stimulation of adrenergic receptors induces an increase in receptor density and is predictive of a decrease in lymphocyte proliferation in response to mitogens [
60]. Interestingly, we also found an upregulation of
ADRB2 after radiation in 1
g as well as in µg, although less pronounced in µg. Ligand-independent receptor activation has been observed for some cellular receptors. For example, radiation-induced activation of the endothelial growth factor receptor (EGFR) has been extensively investigated [
61,
62,
63]. However, whether or not radiation increases receptor density and whether or not the effect, if any, is radiation/gravity-dependent, needs to be investigated. Furthermore, it has been shown that chronic stimulation of adrenergic receptors leads to accumulation of DNA strand breaks by MDM2-mediated degradation of p53 [
64]. Likewise, our results showed a statistically significant increase in
MDM2 and a decrease
TP53 gene expression after 0.8 and 2 Gy of radiation in both 1
g and µg conditions.
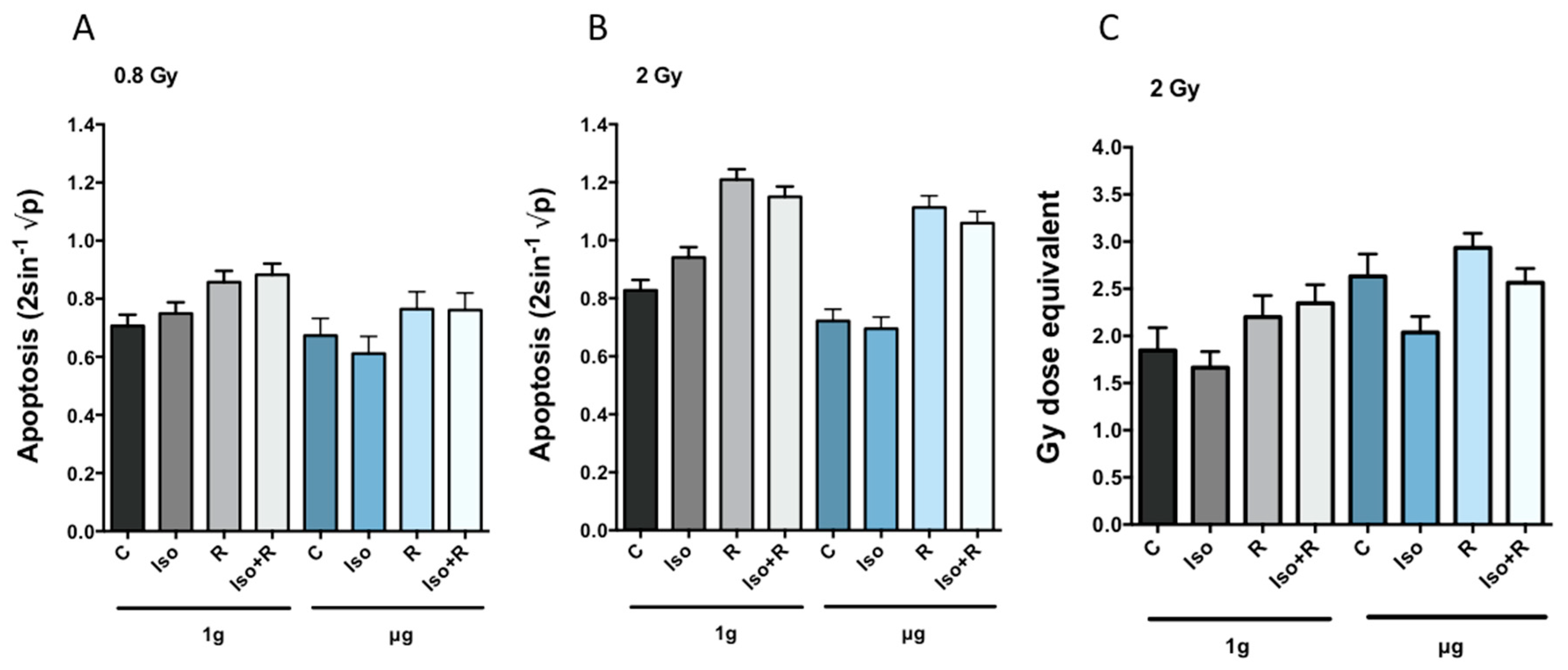
The percentage of PBMCs undergoing radiation-induced apoptosis was similar to previously published data [
65,
66]. Also, in accordance with previous studies [
20,
25], our results indicate a slight reduction in the percentage of apoptotic cells in simulated microgravity alone, although this effect could not be regarded as statistically significant. Furthermore, as published before [
67], we also found that isoproterenol inhibited radiation (2 Gy)-induced apoptosis in cells incubated in 1
g (Iso × R (1
g)) although this was not the case in µg. Isoproterenol-mediated inhibition of radiation-induced apoptosis could be due to an increase in cAMP signaling, which has been shown to inhibit apoptosis by reducing ATM-dependent activation of NF-κB [
68] and by preventing p53 accumulation [
34]. Interestingly, propranolol, a β-adrenergic receptor antagonist, combined with radiation increased apoptosis in human gastric adenocarcinoma cell lines [
69], suggesting an antiapoptotic function of β-adrenergic receptor in irradiated cells. However, without radiation isoproterenol induces apoptosis in 1
g but not in µg leading to a significant interaction between response to isoproterenol and microgravity (Iso × g). It has been shown that stimulation of β-adrenergic receptor induces apoptosis in thymocytes [
33] and the catecholamine dopamine and dobutamine induce apoptosis in peripheral blood mononuclear cells after 24 and 48 h of ex vivo incubation [
67]. The lack of isoproterenol-mediated inhibition of apoptosis in µg could simply reflect the fact of lower apoptotic cells in microgravity. Our results do not support previous findings reporting an impaired apoptosis response to radiation under simulated microgravity [
16,
25]. As mentioned above, most simulated microgravity studies have been conducted using proliferative cells, either cell lines or ex vivo stimulated primary lymphocytes. It is well known that stimulation with growth factors can influence cell death [
70,
71,
72]. Furthermore, stimulated lymphocytes go into a p53-dependent, p21-mediated growth arrest, whereas nonstimulated lymphocytes rapidly go into p53-dependent apoptosis [
73]. Moreover, radiation-induced apoptosis in lymphocytes can be mediated by p53-dependent or p53-independent mechanisms [
74]. We observed a radiation-induced downregulation of p53 in 1
g as well as in µg, which tended to be more pronounced in microgravity (
Figure 3). Interestingly, the ATR-p53 signal pathway is downregulated in quiescent lymphocytes, especially after radiation [
58]. Jones et al. suggest a repressed DNA damage response might protect quiescent lymphocytes from the potential induction of p53-dependent apoptosis despite an endurable DNA damage [
58] and another study shows activation of antiapoptotic genes in the quiescence state [
75]. Interestingly, we found increased amount of DNA strand breaks after 24 h of microgravity exposure suggesting that adaptation to microgravity might be accompanied by accumulation of DNA damage and/or less effective DNA repair mechanisms. No significant synergistic effects were found when the low radiation dose (0.8 Gy) was applied. This could be due to the differences in the responses of different subpopulations to low- and high-dose radiation. In an in vivo mouse study CD8(+) T and B cells were rather resistant to low doses but were very sensitive to 2 Gy, while NK cells, DCs and regulatory T cells (Tregs) cells were much more resistant to high doses [
22].
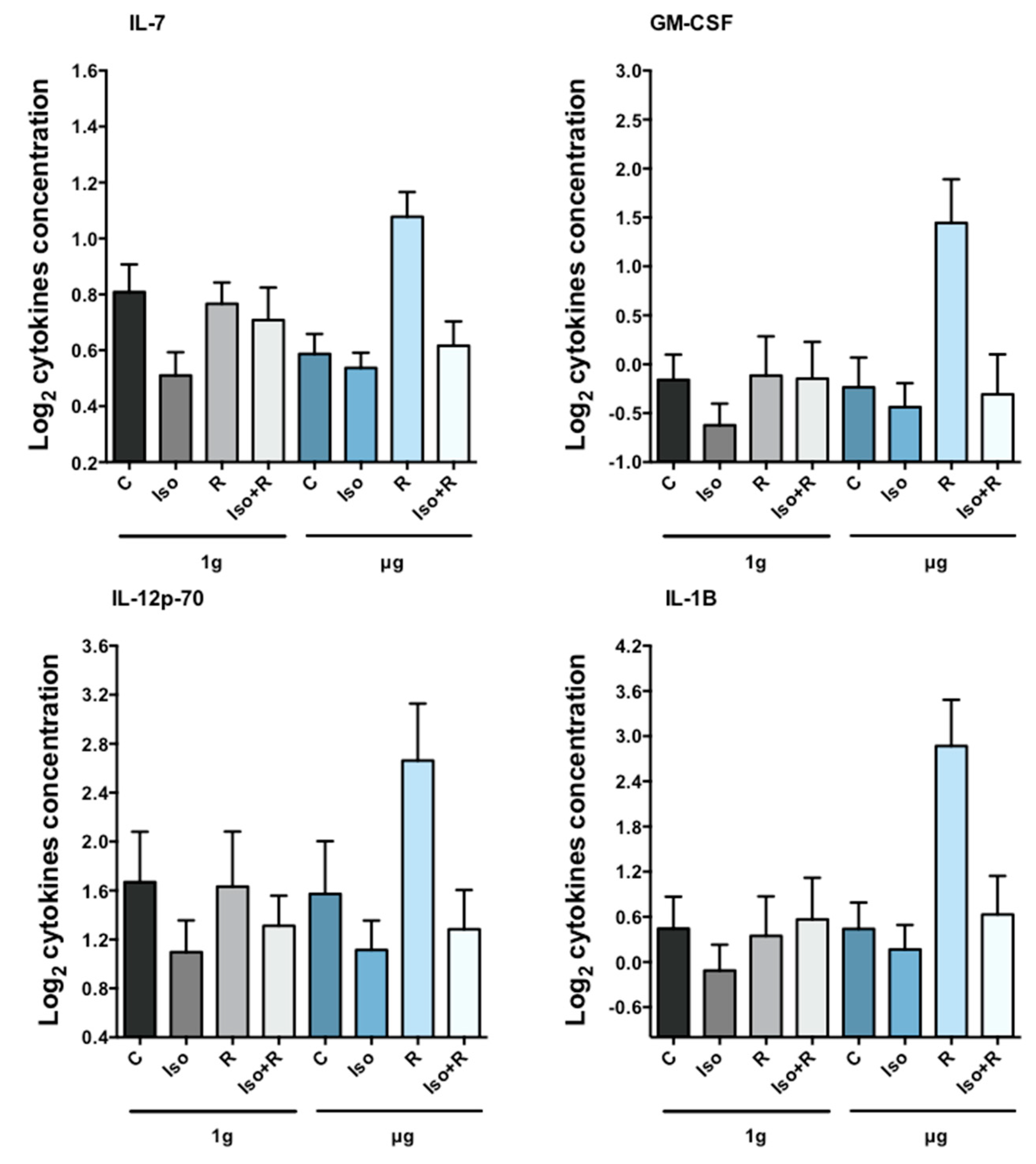
Cytokines are molecules produced in response to stimuli that play a crucial role in regulating cell adhesion, immune recognition, cell death and survival, cell cycle arrest and proliferation, and metabolism [
76]. Ionizing radiation stimulates secretion of pro-inflammatory cytokines in a dose-dependent manner [
77]. Cytokine production in response to low-dose/low-dose rate (Co γ-rays 0.01 Gy, 0.03 cGy/h), with and without acute 2 Gy proton (1 Gy/min) or γ-ray (0.9 Gy/min) irradiation has been investigated also, concluding that the release of at least some cytokines in response to acute 2 Gy radiation is dependent on the radiation quality at the time of assessment, and on the pre-exposure to low-dose radiation with γ-rays [
78]. Isoproterenol suppresses release of cytokines from concanavalin A-activated T cells [
79], and microgravity-induced changes in cytokine release have been reported recently in mitogen-stimulated cells [
80]. However, radiation-induced cytokine production under microgravity conditions has not been investigated. We found increased concentration of some cytokines after radiation but only in cells incubated in microgravity, which was completely abolished by previous treatment with isoproterenol. These findings support and complement in vivo data obtained from mouse hind limb suspension microgravity-models. The combination of 2 Gy (but not low dose) of gamma or proton radiation and hind limb unloading led to an increase in circulating TNFα, while when used separately did not show any effect [
81]. Corresponding to protein levels, we also found a radiation-induced increase in
TNF gene expression only in microgravity (data not shown).
However, cytokine levels relate to cell functionality. There is growing evidence linking DNA damage response elements with inflammatory responses. TGF-β, IL-6, and thrombopoietin influence ATM-dependent DNA damage response, and have been proposed as biomarkers of radioresistance [
82]. Furthermore, NER, responsible for the removal of UV-mediated DNA damage, can be modulated by cytokines, including IL-12, IL-18, and α-melanocyte-stimulating hormone [
83]. In addition, IL-1α can act as an intracellular DNA-damage sensor, signaling cellular genotoxic stress [
84]. A link between the immune system and DNA repair can be found in the V(D)J rearrangement process, responsible for the production of a large repertoire of antigen receptors with different specificities, a requirement for the normal functioning of the immune system. DNA repair proteins such as DNA-PKcs, Ku70, Ku80, XRCC4, LIG4, and Artemis are also involved in the V(D)J recombination [
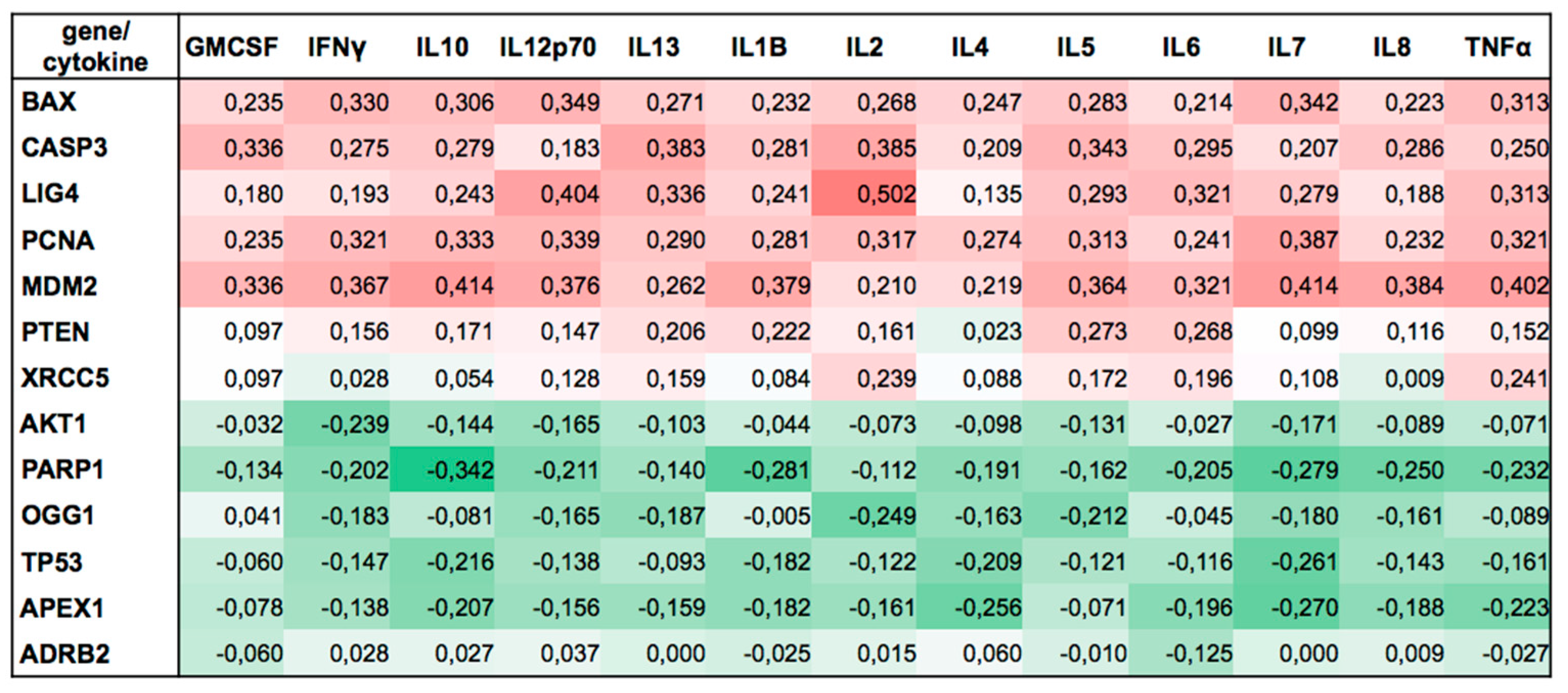
85]. Here, we found associations between intrasubject changes in cytokines and corresponding changes in genes over the 8 experimental conditions. Expressions of
BAX,
CASP3,
PCNA,
LIG4, and
MDM2 were positively correlated with all 13 cytokines studied. Conversely, expressions of
AKT1,
TP53,
PARP1,
OGG1, and
APXE1 were negatively correlated with these cytokines (
Figure 5). Interestingly, for each gene, the sign of the correlation was consistent among all analyzed cytokines. Although it was not the intent of this study to investigate relations between cytokines and DNA damage response, our findings suggest a new link between immune cells and DNA damage response under microgravity conditions. Therefore, we point this out as an interesting starting point for future research necessary for understanding this relationship.
In summary, our results indicate synergistic effects between microgravity, radiation and adrenergic receptor activation. However, our study has some limitations. All measurements were conducted on samples of freshly isolated PBMCs from different subjects. Within dose groups, linear mixed model regression analyses take baseline interindividual differences into account; therefore uncertainties in the estimates of the contrasting effects are due to random interactions between the individuals and the experimental conditions.
The widely recommended housekeeping genes (HKG) ACTB and GAPDH, especially ACTB, turned out to be too dependent on the experimental conditions to provide reliable normalization of expression within PCR plates. Instead, we chose HKG empirically to be ATM, CREB1, and PRKACA using the dual criteria of relatively low dependence on the experimental conditions, while still being sensitive to overall differences between PCR plates. Choosing HKG empirically is a strategy that is gaining acceptance among a wide variety of modern PCR studies. However, given the small number of genes analyzed here, our results need to be confirmed. Nevertheless, the fact that our data confirmed radiation-induced dysregulation of genes involved in DNA damage response in a dose dependent manner supports our strategy as a plausible alternative way for evaluating gene expression analysis.
Of note, our study was exploratory, being designed to identify key elements of the DNA damage response that appeared to be significantly affected by one or more of the mentioned factors. Because of testing multiplicity, even with moderate control of the false-discovery rate, verification of these results through additional controlled studies is recommended.
4. Materials and Methods
4.1. Isolation of PBMCs from Whole Blood
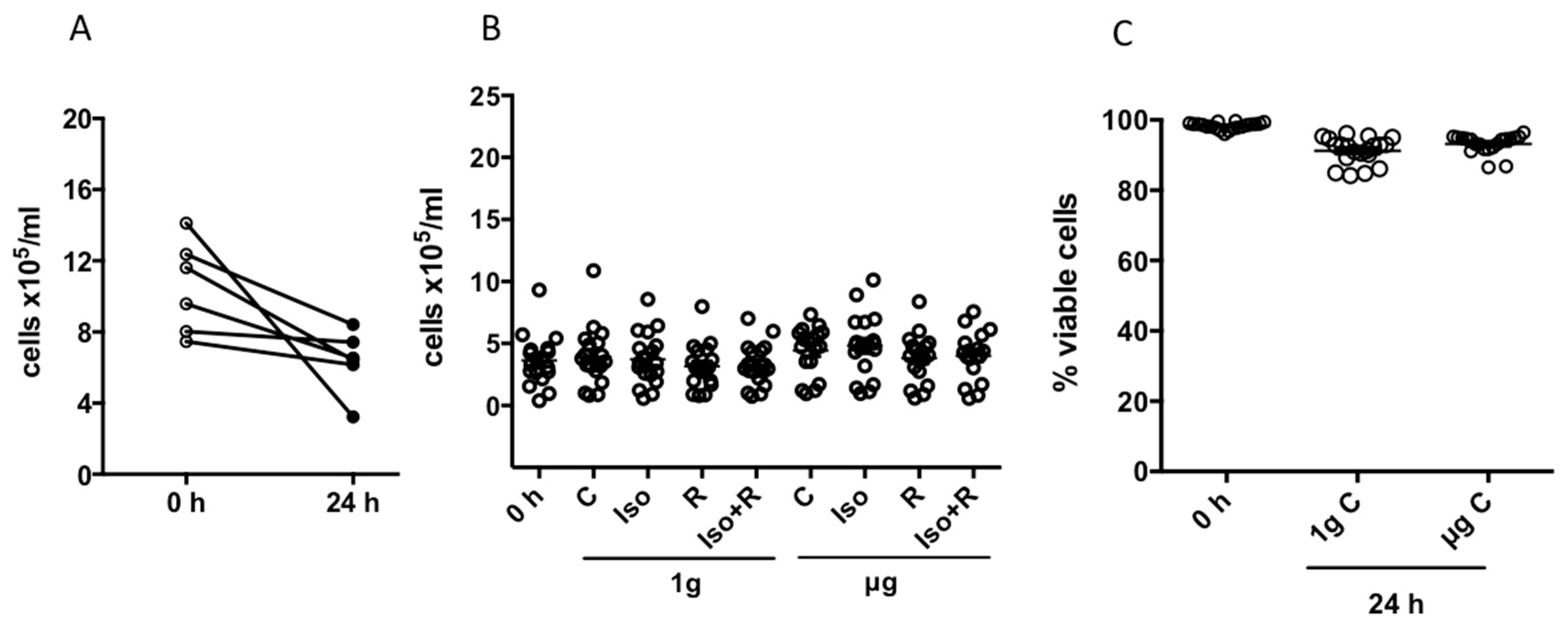
Blood was obtained from volunteers in accordance with accepted ethical and humane practices. Ethical approval was obtained from NASA Johnson Space Center Institutional Review Board, protocol number Pro0614. Whole blood samples were drawn into BD Vacutainer® or CPT™ Mononuclear Cell Preparation Tubes containing sodium heparin (BD Biosciences, Franklin Lakes, NJ, USA). Peripheral blood mononuclear cells (PBMC) were isolated following the manufacturer’s instructions by directly centrifuging the CPT™ to obtain a density gradient or, for the blood collected in the BD Vacutainer® tubes, using the Ficoll-PaqueTM PLUS (GE Healthcare, Uppsala, Sweden). Isolated PBMCs were transferred into 15-mL tubes and washed with phosphate buffered saline (PBS) (Gibco®, Waltham, MA, USA). Tubes were then centrifuged at 300× g for 10 min, the supernatant was removed, and the cell pellet was resuspended in 40 mL of TexMacs medium (Miltenyl Biotec, Auburn, CA, USA). Cells were counted using Guava ViaCount technology (EMD Millipore Co., Hayward, CA, USA). For DNA strand breaks analysis, isolated cells were suspended in 1 mL of freezing medium containing 20% Roswell Park Memorial Institute medium (RPMI-1640) medium, 10% dimethyl sulfoxide (DMSO), and 70% fetal calf serum (FCS), and stored overnight at −80 °C in a Mr. Frosty™ Freezing Container (Thermo Fisher Scientific, USA). The cells were then transferred to a liquid nitrogen tank at −180 °C until shipment to Konstanz (Germany), where cells were kept at −180 °C until analysis. Then, PBMCs were carefully thawed by immersing cryovials in a water bath at 37 °C until a small amount of ice remained in the cryovial and thereafter adding 1 mL of thawing medium (90% RPMI and 10% FCS). After 1 min the cell suspension was transferred into a polypropylene 15 mL tube and thawing medium was added stepwise (1 mL, 1 min later additional 2 mL, 1 min later additional 4 mL were added). The tube was centrifuged at 300× g for 10 min. The cell pellet was gently resuspended in 1 mL RPMI medium, and the cell concentration and viability (determined by electric current exclusion) were assessed using CASY cell counter technology (Innovatis, Switzerland). No significant difference was detected in the cell vitality of control cells after thawing (68.2 ± 7% and 65.2 ± 5.6% in simulated microgravity and 1g, respectively).
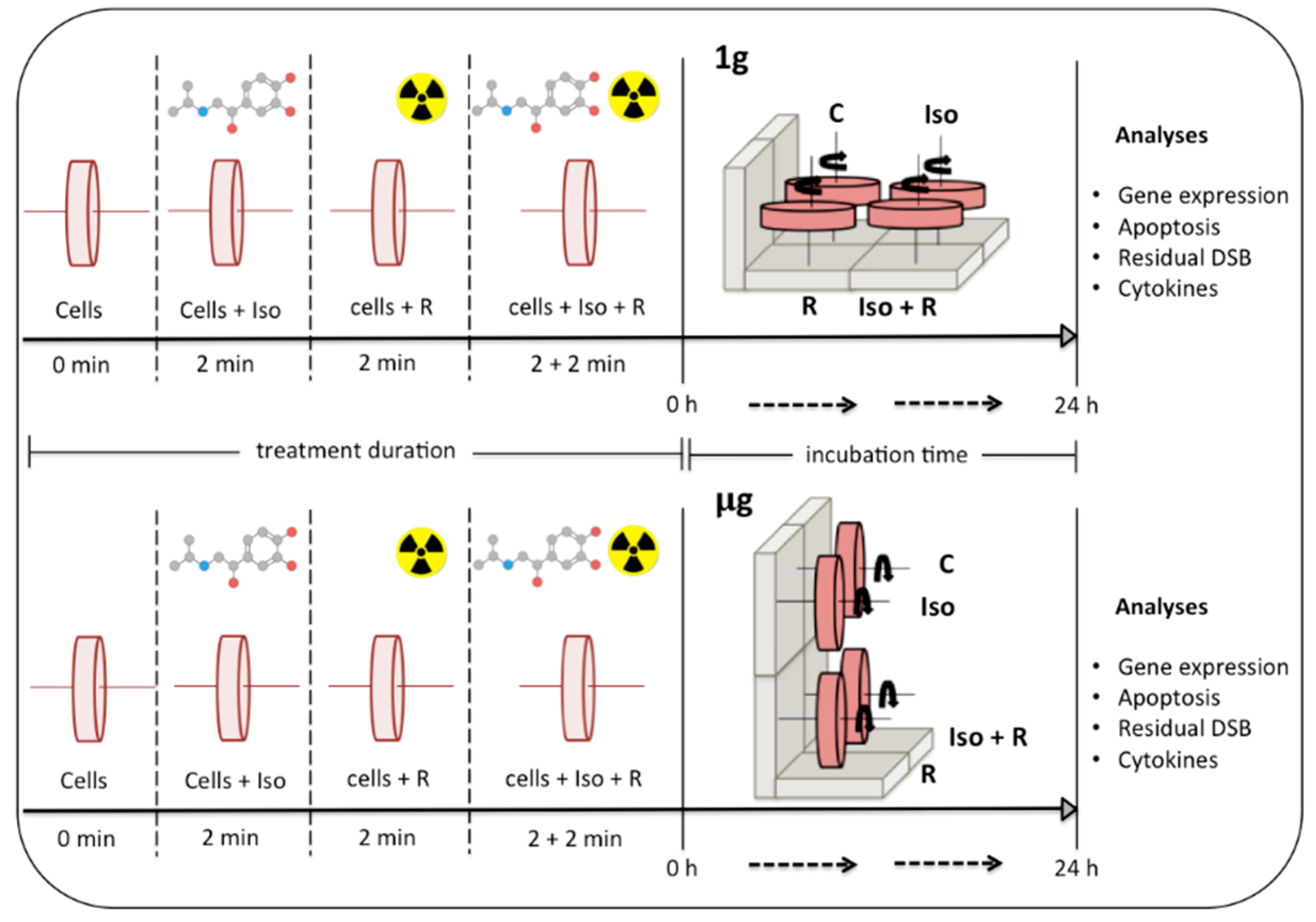
4.2. Experimental Design
To mimic the absence of gravity, cell suspensions were added to rotating cell culture system vessels (RCCSVs) (Synthecon Inc., Houston, TX, USA) similar to the RWVs originally developed by NASA. Four vessels were allocated to Earth gravity experimental conditions (1
g) and were rotated horizontally, while four others were allocated to simulated microgravity conditions (µg) and were rotated vertically. For each level of gravity (1
g and µg), cells in vessels were either, (1) not treated (control), (2) treated with 10µM (-)-isoproterenol hydrochloride (Sigma-Aldrich, Milwaukee, WI, USA), (3) irradiated (0.8 or 2 Gy), or (4) treated with 10µM (-)-isoproterenol hydrochloride and immediately irradiated (0.8 or 2 Gy) (
Figure 6). Vessels were placed on the rotary cell culture systems (Synthecon Inc., Houston, TX, USA). After treatment and/or radiation all vessels rotated synchronously at a speed of 8.5 rpm for 24 h in an incubator (37 °C, 5% CO
2, and 95% relative humidity). After incubation, cells were recovered from the RCCSVs and cell concentration and viability was determined using Guava ViaCount technology (EMD Millipore, Hayward, CA, USA) prior to further analyses.
4.3. Gene Expression
After incubation, cells were centrifuged at 300× g for 10 min after which the supernatant fluid was carefully removed. RNA was isolated from each cell pellet using AllPrep DNA/RNA/miRNA Universal Kit (Quiagen, Hilden, Germany). RNA concentration was measured using a NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription was performed using the miScript II RT Kit (Quiagen, Hilden, Germany), and conversion to cDNA was performed in the DNA Engine® Thermal Cycler (BioRad, Hercules, CA, USA). cDNA was diluted in 40 µL RNase-free water. The PCR reagent consisted of 12.5 µL 2xQuantiTect SYBR Green PCR Master Mix, 7.5 µL RNase-free water, 2.5 µL 10x Primer Assay, and 2.5 µL Template cDNA. PCR was performed in a CFX96TM Thermal Cycler (BioRad, Hercules, CA, USA). Genes of interest were selected according to the following criteria: (i) genes that are major players and representative of the pathways described in Secs 1.1 and 1.2); (ii) genes that are known to be involved in two or more of these relevant suggested pathways; (iii) genes that are expected to undergo adaptive responses; (iv) genes coding a considerable number of proteins known to be regulated at the mRNA level.
4.4. Apoptosis
The percentage of dead cells after incubation was determined by diluting samples 1:1 with Guava Nexin® reagent (EMD Millipore Co., Billerica, MA, USA). These mixtures were incubated for twenty minutes. Apoptotic cells were counted in Guava-PCA machine (EMD Millipore, Hayward, CA, USA).
4.5. Detection of DNA Strand Breaks
DNA strand breaks were detected using the automated version of the “Fluorimetric detection of Alkaline DNA Unwinding” (FADU) assay [
86,
87]. This assay is based on controlled DNA unwinding that starts at DNA strand breaks. SybrGreen
® (MoBiTec, Göttingen, Germany) was used as the marker for double stranded DNA. A decrease in the fluorescence intensity indicates an increase in DNA unwinding and consequently a greater number of strand breaks. The fluorescence signal was transformed into equivalent radiation dose units (Gy) [
88].
4.6. Cytokine Quantification
Medium supernatant was used for cytokine measurements. The concentrations for 13 cytokines were determined simultaneously in a 96-well plate in duplicate using a commercially available multiplex bead immunoassay (R&D Systems). Briefly, 50 µL of medium supernatant were incubated with 13 sets of beads each precoated with specific antibodies against cytokines of interest. The bead sets fluoresce at different wavelengths so that individual cytokines can be identified. After a washing step, the bead–cytokine complex was incubated with fluorescence secondary antibodies specific for each cytokine that fluoresce along a single channel distinct from the bead populations. Fluorescence, indicating relative concentration, was assessed using a Luminex 100 instrument (Luminex, Inc. Austin, TX, USA). Cytokines were selected based on their different types of biological responses including innate immunity, adaptive immunity, and growth factors, and based on their physiological functions: pro- versus anti-inflammatory properties.
4.7. Statistical Analysis
Each blood sample was split into eight subsamples following a repeated-measures 2
3-factorial design with radiation (yes, no), isoproterenol (yes, no), and simulated microgravity (yes, no) as factors (
Figure 6). For all analyses (gene expression, apoptosis, cytokines, and DNA strand breaks), mixed model regression with bootstrapped standard errors (200 reps) was used to make inference on contrasts involving the experimental factors. Mixed model random effects were modeled at the sample level (apoptosis, cytokines, strand breaks) and at the PCR-plate level (gene expression). To account for test multiplicity, we used either the method of Benjamini Y., A. Krieger, and D. Yekutieli [
89] to control the false-discovery rate (FDR) or the method of Holm [
90] to control the family-wise Type I error rate (FWER) depending on the number of tests run. A summary of statistical methods applied for each cellular parameter is indicated in
Table 6. The nomenclature for experimental factors and analyzed contrasts is elucidated in
Table 7.
4.8. Comparison of Cytokine and Gene Expression Data
In addition to the separate analyses for each cellular parameter, we also used the Somers D [
91,
92] statistic to create the table of associations (
Figure 5) between within-subject changes in cytokines and corresponding changes in genes over the eight experimental conditions.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}