Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
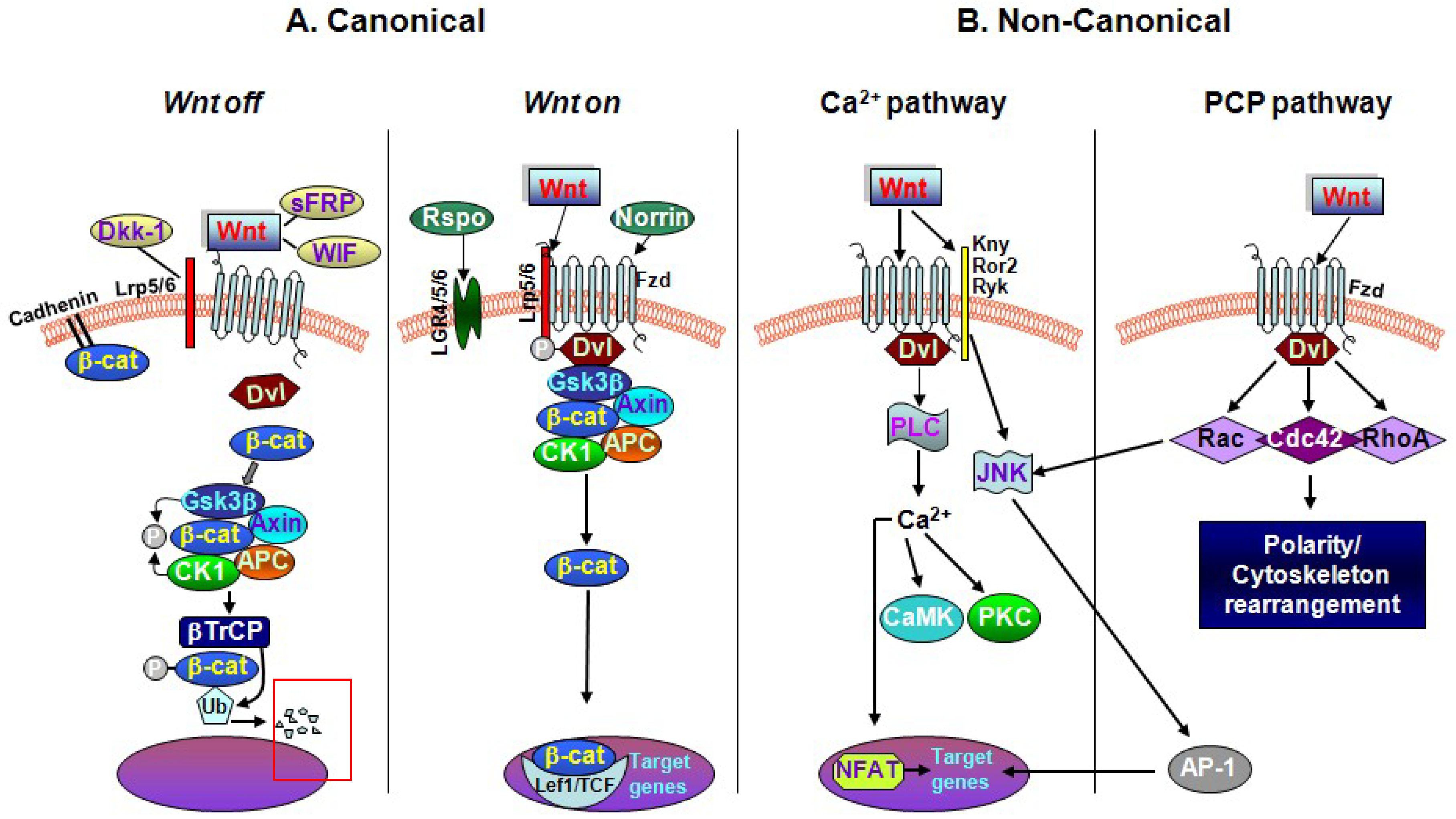
2. The Key Interactors of the Wnt/β-catenin Signaling Cascade
3. The MPTP Mouse Model Highlights Neuroinflammation as a Critical Risk Factor for PD
4. Dysregulated Wnt/β-catenin Signaling Is Linked to Gene Defects in PD
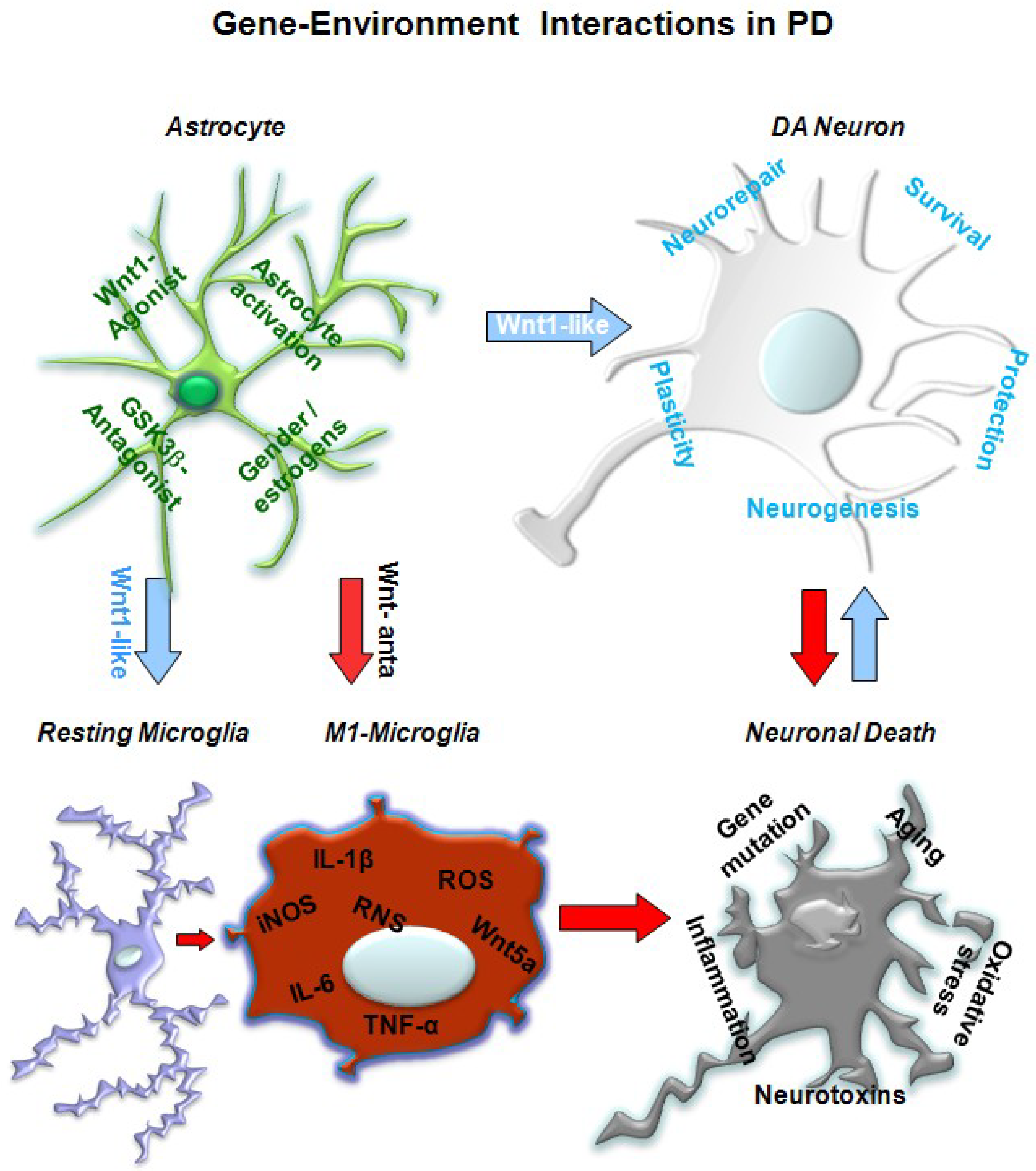
5. A Wnt1 Self-Protective Regulatory Loop Engaged by Astrocyte–Neuron Cross-talk Safeguards mDAergic Neurons
5.1. In Vivo Studies Uncover Wnt Signaling Intermediacy as Chief Player in Glial Modulation
5.2. In Vitro Studies Pinpoint Wnt1/Fzd1/β-catenin Survival Pathway for mDAergic Neurons
6. Deficient Wnt Signaling with Age: A Leading Risk Factor for Nigrostriatal DAergic Neuron Integrity
7. Wnt/β-catenin Signaling Is Required for DAergic Neurorepair and Regeneration
8. Targeting Wnt Signaling as a “Wn(t)dow” of Opportunity for mDAergic Neurorescue
9. Concluding Remarks and Future Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| Dkk | Dkkopf |
| DA | Dopamine |
| DAergic | Dopaminergic |
| DAT | DA transporter |
| Fzd | Frizzled |
| GFAP | glial fibrillary acid protein |
| GSK-3β | glycogen synthase kinase-3β |
| iNOS | inducible nitric oxide |
| IFN-g | interferon-g |
| IL-1a | interleukin-1a |
| IL-1b | interleukin-1b |
| LRP5/6 | LDL receptor-related protein 5/6 |
| LRRK2 | leucine-rich repeat kinase 2 |
| MB | midbrain/hindbrain |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropiridine |
| MPP+ | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NO | nitric oxide |
| Phox | NADPH oxidase |
| ONOO− | peroxynitrite |
| PD | Parkinson’s disease |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| 6-OHDA | 6-hydroxydopamine |
| NSC | stem/neuroprogenitor cell |
| SNpc | subtantia nigra pars compacta |
| SVZ | subventricular zone |
| TH | tyrosine hdroxylase |
| TNF-a | tumor necrosis factor a |
| VM | ventral midbrain |
| Wnt1 | Wingless-type MMTV integration site 1 |
References
- Del Tredici, K.; Braak, H. Lewy pathology and neurodegeneration in premotor Parkinson’s disease. Mov. Disord. 2012, 27, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Shapira, A.H.V. Therapeutic prospects for Parkinson’s disease. Ann. Neurol. 2013, 74, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. Gene-environment interactions in Parkinson’s disease: Specific evidence in humans and mammalian models. Neurobiol. Dis. 2013, 57, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.A.; Langston, J.W. Idiopathic and 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. In Neuroglia; Kettenmann, H., Ransom, B.R., Eds.; Oxford University Press: Oxford, UK, 1995; pp. 997–998. [Google Scholar]
- Warner, T.T.; Schapira, A.H.V. Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 2003, 53, S16–S25. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP Story. J. Parkinsons Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.-M.; Hong, J.-S. Gene–environment interactions: Key to unraveling the mystery of Parkinson’s disease. Prog. Neurobiol. 2011, 94, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C. Vulnerability to Parkinson’s Disease: Towards an Unifying Theory of Disease Etiology. In Encyclopedia of Environmental Health; Jerome, O.N., Ed.; Elsevier Science: Oxford, UK, 2011; pp. 690–704. [Google Scholar]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell Probes 2016, 30, 386–396. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Fernández, R.; Schulzer, M.; Kuramoto, L.; Cragg, J.; Ramachandiran, N.; Au, W.L.; Mak, E.; McKenzie, J.; Mc Cormick, S.; Sossi, V.; et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann. Neurol. 2011, 69, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s disease as a result of aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; Abbracchio, M.P. To be or not to be (inflammed) is that the question in anti-inflammatory drug therapy of neurodegenerative diseases? Trends Pharmacol. Sci. 2005, 26, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jacobs, E.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Non steroidal anti-inflammatory drugs and the risk of Parkinson’s disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; Serra, PA.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Cioni, S.; Gennuso, F.; Rocchitta, G.; Desole, M.S.; et al. Hormones are key actors in gene x environment interactions programming the vulnerability to Parkinson’s disease: Glia as a common final pathway. Ann. N. Y. Acad. Sci. 2005, 1057, 296–318. [Google Scholar] [CrossRef] [PubMed]
- Morale, M.C.; Serra, P.; Delogu, M.R.; Migheli, R.; Rocchitta, G.; Tirolo, C.; Caniglia, S.; Testa, N.; L’Episcopo, F.; Gennuso, F.; et al. Glucocorticoid receptor deficiency increases vulnerability of the nigrostriatal dopaminergic system: Critical role of glial nitric oxide. FASEB J. 2004, 18, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Morale, M.C.; L’Episcopo, F.; Tirolo, C.; Giaquinta, G.; Caniglia, S.; Testa, N.; Arcieri, P.; Serra, P.A.; Lupo, G.; Alberghina, M.; et al. Loss of Aromatase Cytochrome P450 function as a risk factor for Parkinson’s disease? Brain Res. Rev. 2008, 57, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S. Inflammation and Parkinson’s disease pathogenesis. Mov. Disord. 2010, 25, S55–S57. [Google Scholar] [CrossRef] [PubMed]
- Morale, M.C.; Serra, P.A.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Gennuso, F.; Giaquinta, G.; Rocchitta, G.; Desole, M.S.; et al. Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: Glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 2006, 138, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Abou-Sleiman, P.M.; Muqit, M.M.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 365–3668. [Google Scholar] [CrossRef] [PubMed]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front Neurol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Wurst, W.; Prakash, N. Wnt-1 regulated genetic networks in midbrain dopaminergic neuron development. J. Mol. Cell Biol. 2014, 6, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E. Wnt signaling in midbrain dopaminergic neuron development and regenerative medicine for Parkinson’s disease. J. Mol. Cell Biol. 2014, 6, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, M.; Awatramani, R. Wnt/β-catenin signaling in midbrain dopaminergic neuron specification and neurogenesis. J. Mol. Cell Biol. 2014, 6, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Parish, C.L.; Thompson, L.H. Modulating Wnt signaling to improve cell replacement therapy for Parkinson’s disease. J. Mol. Cell Biol. 2014, 6, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Toledo, E.M.; Gyllborg, D.; Arenas, E. Translation of WNT developmental programs into stem cell replacement strategies for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2017, 174, 4716–4724. [Google Scholar] [CrossRef] [PubMed]
- Salinas, P.C. Wnt signaling in the vertebrate central nervous system: From axon guidance to synaptic function. Cold Spring Harb. Perspect Biol. 2012, 4, A008003. [Google Scholar] [CrossRef]
- Grainger, S.; Willert, K. Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, e1422. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt Signaling. Cold Spring Harb. Perspect Biol. 2012, 4, A011163. [Google Scholar] [CrossRef] [PubMed]
- Kalani, M.Y.; Cheshier, S.H.; Cord, B.J.; Bababeygy, S.R.; Vogel, H.; Weissman, I.L.; Palmer, T.D.; Nusse, R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16970–16975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Nusse, R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R. Alternative wnt pathways and receptors. Cold Spring Harb. Perspect Biol. 2012, 4, A007914. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Clevers, H. WNT signalling events near the cell membrane and their pharmacological targeting for the treatment of cancer. Br. J. Pharmacol. 2017, 174, 4547–4563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiese, K.; Li, F.; Chong, Z.Z.; Shang, Y.C. The Wnt signalling pathway: Aging gracefully as a protectionist? Pharmacol. Ther. 2008, 118, 58–81. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Arenas, E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010, 11, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt Signalling in the Nervous System and in Alzheimer’s Disease. In “Wnt signaling cascades in neurodevelopment, neurodegeneration and regeneration”. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Galli, S.; Salinas, P.C. Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. In “Wnt signaling cascades in neurodevelopment, neurodegeneration and regeneration”. J. Mol. Cell Biol. 2014, 6, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.; et al. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 regulated Frizzled-1/β-catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011, 13, 6–49. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; Pluchino, S. Wnt your brain be inflamed? Yes, it Wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Morale, M.C.; Serapide, M.F.; Pluchino, S.; Marchetti, B. Targeting Wnt signaling at the neuroimmune interface in dopaminergic neuroprotection/repair in Parkinson’s disease. J. Mol. Cell Biol. 2014, 6, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; L’Episcopo, F.; Morale, M.C.; Tirolo, C.; Testa, N.; Caniglia, S.; Serapide, M.F.; Pluchino, S. Uncovering novel actors in astrocyte-neuron crosstalk in Parkinson’s disease: The Wnt/β-catenin signaling cascade as the common final pathway for neuroprotection and self-repair. Eur. J. Neurosci. 2013, 37, 1550–1563. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Serapide, M.F.; Deleidi, M.; Pluchino, S.; Marchetti, B. Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson’s disease involves crosstalk between inflammatory and Wnt/β-catenin signaling pathways: Functional consequences for neuroprotection and repair. J. Neurosci. 2012, 32, 2062–2085. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Impagnatiello, F.; Pluchino, S.; Marchetti, B. Aging-induced Nrf2-ARE pathway disruption in the subventricular zone (SVZ) drives neurogenic impairment in parkinsonian mice via PI3K-Wnt/β-catenin dysregulation. J. Neurosci. 2013, 33, 1462–1485. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Serapide, M.F.; Caniglia, S.; Testa, N.; Leggio, L.; Vivarelli, S.; Iraci, N.; Pluchino, S.; Marchetti, B. Microglia polarization, gene-environment interactions and Wnt/β-catenin Signalling: Emerging roles of glia-neuron and glia-stem/neuroprogenitor crosstalk for dopaminergic neurorestoration in aged parkinsonian brain. Front. Aging Neurosci. 2018, 10, 12. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Peruzzotti-Jametti, L.; Serapide, M.F.; Testa, N.; Caniglia, S.; Balzarotti, B.; Pluchino, S.; Marchetti, B. Neural Stem Cell Grafts Promote Astroglia-Driven Neurorestoration in the Aged Parkinsonian Brain via Wnt/β-Catenin Signaling. Stem Cells 2018, 36, 1179–1197. [Google Scholar] [CrossRef]
- Gollamudi, S.; Johri, A.; Calingasan, N.Y.; Yang, L.; Elemento, O.; Beal, M.F. Concordant signaling pathways produced by pesticide exposure in mice correspond to pathways identified in human Parkinson’s disease. PLoS ONE 2012, 7, e36191. [Google Scholar] [CrossRef] [PubMed]
- Ohnuki, T.; Nakamura, A.; Okuyama, S.; Nakamura, S. Gene expression profiling reveals molecular pathways associated with sporadic Parkinson’s disease. Brain Res. 2010, 1346, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, I.; Keller-Mccgandy, C.; Bouziou, B.; Asteris, G.; Clark, T.W.; Frosh, M.P.; Standaert, D.G. Effects of gender on nigral gene expression and parkinson’s disease. Neurobiol. Dis. 2007, 26, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.B.; Hallupp, M.; Loy, C.T.; Chan, D.K.; Woo, J.; Mellick, G.D.; Buchanan, D.D.; Silburn, P.A.; Halliday, G.M.; Schofield, P.R. GSK3β polymorphism alter transcription and splicing in Parkinson’s disease. Ann. Neurol. 2005, 58, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Deng, J.; Pan, Q.; Zhan, Y.; Fan, J.B.; Zhang, K.; Zhang, Z. Targeted methylation sequencing reveals dysregulated Wnt signaling in Parkinson disease. J. Genet. Genom. 2016, 43, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, O.; Liu, Q.; Swistowski, A.; Russo-Tait, T.; Zhao, Y.; Rao, M.S.; Zeng, X. Genome wide profiling of dopaminergic neurons derived from human embryonic and induced pluripotent stem cells. Stem Cells Dev. 2014, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Moya, N.; Cutts, J.; Gaasterland, T.; Willert, K.; Brafman, D.A. Endogenous WNT signaling regulates hPSC-derived neural progenitor cell heterogeneity and specifies their regional identity. Stem Cell Rep. 2014, 3, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Harvey, K. The regulation and deregulation of Wnt signalling by PARK genes in health and disease. J. Mol. Cell Biol. 2014, 6, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Javaheri, B.; Wetzel, A.; Hopkinson, M.; Nixon-Abell, J.; Grannò, S.; Pitsillides, A.A.; Harvey, K. Pathogenic LRRK2 variants are gain-of-function mutations that enhance LRRK2-mediated repression of β-catenin signaling. Mol. Neurodegener. 2017, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Nixon-Abell, J.; Berwick, D.C.; Grannó, S.; Spain, V.A.; Blackstone, C.; Harvey, K. Protective LRRK2 R1398H Variant Enhances GTPase and Wnt Signaling Activity. Front. Mol. Neurosci. 2016, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Panicker, L.M.; Deranieh, R.M.; Srikanth, M.P.; Brown, R.A.; Voit, A.; Peesay, T.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Altered differentiation potential of Gaucher’s disease iPSC neuronal progenitors due to Wnt/β-catenin downregulation. Stem Cell Rep. 2017, 9, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, F.; Zhu, G.; Feng, D.; Nie, T.; Tao, K.; Yang, S.; Lei, J.; Huang, L.; Mao, Z.; et al. Loss of Drosha underlies dopaminergic neuron toxicity in models of Parkinson’s disease. Cell Death Dis. 2018, 9, 693. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, H.; Song, H.; Qin, Y.; Wang, Y.; Xu, M.; Liu, C.; Gao, J.; Sun, S. Let-7d microRNA Attenuates 6-OHDA-Induced Injury by Targeting Caspase-3 in MN9D Cells. J. Mol. Neurosci. 2017, 63, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Leggio, L.; Vivarelli, S.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Marchetti, B.; Iraci, N. microRNAs in Parkinson’s Disease: From Pathogenesis to Novel Diagnostic and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 2698. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Botta-Orfila, T.; Moratò, X.; Calatayud, C.; Ferrer-Lorente, R.; Marti, M.J.; Fernández, M.; Gaig, C.; Raya, Á.; Consiglio, A.; et al. MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. Neurobiol. Aging 2018, 69, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Song, J.L.; Nigam, P.; Tektas, S.S.; Selva, E. microRNA regulation of Wnt signaling pathways in development and disease. Cell Signal. 2015, 27, 1380–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, S.; Bhatti, A.; Syed, N.A.; John, P. The microRNA regulatory network: A far-reaching approach to the regulate the Wnt signaling pathway in number of diseases. J. Recept Signal Transduct. 2016, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gotz, S.; Vogt Weisenhorn, D.M.; Simeone, A.; Wurst, W.; Prakash, N. A WNT1-regulated developmental gene cascade prevents dopaminergic neurodegeneration in adult En1(+/-) mice. Neurobiol. Dis. 2015, 82, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.; Lopes, D.M.; Ammari, R.; Kopra, J.; Millar, S.E.; Gibb, A.; Salinas, P.C. Deficient Wnt signalling triggers striatal synaptic degeneration and impaired motor behaviour in adult mice. Nat. Commun. 2014, 5, 4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Ruiz, O.; Zhang, Y.J.; Shan, F.; Malik, N.; Hoffman, A.F.; Ladenheim, B.; Cadet, J.L.; Lupica, C.R.; Tagliaferro, A.; Brusco, A.; et al. Attenuated response to methamphetamine sensitization and deficits in motor learning and memory after selective deletion of β-catenin in dopamine neurons. Learn Mem. 2012, 19, 341–350. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Serapide, M.F.; Pluchino, S.; Marchett, B. Wnt/β-catenin signaling is required to rescue midbrain dopaminergic progenitors and restore nigrostriatal plasticity in ageing mouse model of Parkinson’s disease. Stem Cells 2014, 32, 2147–2163. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.; Marchetti, B. Regulating Wnt signaling: A strategy to prevent neurodegeneration and induce regeneration. J. Mol. Cell Biol. 2014, 6, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Dun, Y.; Li, G.; Yang, Y.; Xiong, Z.; Feng, M.; Wang, M.; Zhang, Y.; Xiang, J.; Ma, R. Inhibition of the canonical Wnt pathway by Dickkopf-1 contributes to the neurodegeneration in 6-OHDA-lesioned rats. Neurosci. Lett. 2012, 525, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, C.; Lei, M.; Li, G.; Yi, L.; Luo, F.; Li, Y.; Ding, L.; Liu, Z.; Li, S.; et al. Activation of Wnt/β-catenin pathway by exogenous Wnt1 protects SH-SIY cells against 6-hydroxydopamine toxicity. J. Mol. Neurosci. 2013, 49, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Q.; Xi, J.; Xiao, B.; Li, Y.; Ma, C. Neuroprotective effect of fasudil on inflammation through PI3K/Akt and Wnt/β-catenin dependent pathways in a mice model of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 2354–2364. [Google Scholar] [PubMed]
- Zhang, L.; Cen, L.; Qu, S.; Wei, L.; Mo, M.; Feng, J.; Sun, C.; Xiao, Y.; Luo, Q.; Li, S.; et al. Enhancing Beta-Catenin Activity via GSK3beta Inhibition Protects PC12 Cells against Rotenone Toxicity through Nurr1 Induction. PLoS ONE 2016, 11, e0152931. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zu, G.; Zhang, X.; Wang, X.; Li, S.; Gong, X.; Liang, Z.; Zhao, J. Neuroprotective effects of ginsenoside Rg1 through the Wnt/β-catenin signaling pathway in both in vivo and in vitro models of Parkinson’s disease. Neuropharmacology 2016, 101, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Shen, Z.; Yuan, Y.; Han, D.; Song, C.; Guo, Y.; Mei, X. Simvastatin inhibits neural cell apoptosis and promotes locomotor recovery via activation of Wnt/β-catenin signaling pathway after spinal cord injury. J. Neurochem. 2016, 138, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gu, J.; Wu, H.; Zhu, G.; Feng, D.; Li, Y.; Guo, W.; Tian, K.; Gao, G.; Gao, L. Pentazocine Protects SN4741 Cells Against MPP+-Induced Cell Damage via Up-Regulation of the Canonical Wnt/β-Catenin Signaling Pathway. Front. Aging Neurosci. 2017, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hao, S.; Yang, B.; Fan, Y.; Qin, X.; Chen, Y.; Hu, J. Wnt/β-catenin signaling plays an essential role in α7 nicotinic receptor-mediated neuroprotection of dopamnergic neurons in a mouse model of Parkinson’s disease. Biochem. Pharmacol. 2017, 140, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Ju, B.; Zhang, Y.Z.; Yin, H.L.; Liu, Y.J.; Wang, S.S.; Zeng, Z.L.; Yang, X.P.; Wang, H.T.; Li, J.F. Protective Effect of Curcumin Against Oxidative Stress-Induced Injury in Rats with Parkinson’s Disease Through the Wnt/β-Catenin Signaling Pathway. Cell Physiol. Biochem. 2017, 43, 2226–2241. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Mishra, A.; Shukla, S. ALCAR Exerts Neuroprotective and Pro-Neurogenic Effects by Inhibition of Glial Activation and Oxidative Stress via Activation of the Wnt/β-Catenin Signaling in Parkinsonian Rats. Mol. Neurobiol. 2016, 53, 4286–4301. [Google Scholar] [CrossRef] [PubMed]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural basis of Wnt recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Salašová, A.; Yokota, C.; Potesil, D.; Zdráhal, Z.; Bryja, V.; Arenas, E. A proteomic analysis of LRRK2 binding partners reveals interactions with multiple signaling components of the WNT/PCP pathway. Mol. Neurodegener. 2017, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’Episcopo, F.; Drouin-Ouellet, J.; Tirolo, C.; Pulvirenti, A.; Giugno, R.; Testa, N.; Caniglia, S.; Serapide, M.F.; Cisbani, G.; Barker, R.A.; et al. GSK-3β-induced Tau pathology drives hippocampal neuronal cell death in Huntington’s disease: Involvement of astrocyte-neuron interactions. Cell Death Dis. 2016, 7, e2206. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.D.; Olanow, C.W. Differential modulation of Akt/Glycogen synthase kinase-3beta pathway regulates apoptotic and cytoprotective signalling responses. J. Biol. Chem. 2008, 283, 15469–15478. [Google Scholar] [CrossRef] [PubMed]
- Petit-Paitel, A.; Brau, F.; Cazareth, J.; Chabry, J. Involment of cytosolic and mitochondrial GSK-3beta in mitochondrial dysfunction and neuronal cell death of MPTP/Mpp+- treated neurons. PLoS ONE 2009, 4, e5491. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Miyamoto, Y.; Huang, E.J. Multiple roles of β-catenin in controlling the neurogenic niche for midbrain dopamine neurons. Development 2009, 136, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Rawal, N.; Corti, O.; Sacchetti, P.; Ardilla-Osorio, H.; Sehat, B.; Brice, A.; Arenas, E. Parkin protects dopaminergic neurons from excessive Wnt/beta-cateninsignaling. Biochem. Biophys. Res. Commun. 2009, 388, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, P.; Esteve, P.; Ruiz, J.M.; Cisneros, E.; Lopez-Rios, J. Beyond Wnt inhibition: New functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 2008, 121, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Esteve, P.; Sandonìs, A.; Ibanez, C.; Shimono, A.; Guerrero, I.; Bovolenta, P. Secreted frizzled related proteins are required for Wnt/β-catenin signaling activation in the vertebrate optic cup. Development 2011, 138, 4179–4184. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reevers, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP model of Parkinson’s disease. Nat. Prot. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Przedborski, S. A tale on animal models of Parkinson’s Disease. Mov. Disord. 2011, 26, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Marchetti, B. Glia as a turning point in the therapeutic strategy of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 349–372. [Google Scholar]
- Branch, S.Y.; Sharma, R.; Beckstead, MJ. Aging decreases L-Type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J. Neurosci. 2014, 34, 9310–9318. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Lipton, J.; Daley, B.F.; Palfi, S.; Chu, Y.; Sortwell, C.; Bakay, R.A.; Sladek, J.R., Jr.; Kordower, J.H. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: Diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol. Dis. 2007, 26, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezard, E.; Gross, C.E. Compensatory mechanisms in experimental and human parkinsonism: Towards a dynamic approach. Prog. Neurobiol. 1998, 55, 93–116. [Google Scholar] [CrossRef]
- Zigmond, M.J. Do compensatory processes underlie the preclinical phase of neurodegenerative diseases? Insight from an animal model of parkinsonism. Neurobiol. Dis. 1997, 4, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.T.; Chen, X.; Moore, D.J. VPS35, the Retromer Complex and Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Belenkaya, T.Y.; Wu, Y.; Tang, X.; Zhou, B.; Cheng, L.; Sharma, Y.V.; Yan, D.; Selva, E.M.; Lin, X. The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev. Cell. 2008, 14, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Michaux, G.; Le Borgne, R. Sorting, recycling and WNT signaling: Wntless and retromer functions. Med. Sci. (Paris) 2009, 25, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Swan, M.; Saunders-Pullman, R. The association between b-glucocerebrosidase mutations and parkinsonism. Curr. Neurol. Neurosci. Rep. 2013, 13, 368. [Google Scholar] [CrossRef]
- Cox, T.M. Gaucher disease: Clinical profile and therapeutic developments. Biologics 2010, 4, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPISC derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Vitner, E.B.; Kelly, S.L.; Bame, J.R.; Duan, J.; Shinder, V.; Merrill, A.H., Jr.; Dobrenis, K.; Futerman, A.H. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2014, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, R.; Vick, P.; Ploper, D.; Gumper, I.; Snitkin, H.; Sabatini, D.D.; De Robertis, E.M. Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2012, 2, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.; Geczy, C.L.; Halliday, G.M. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience 2015, 302, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2-deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Human Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.F.; Ho, P.W.; Leung, G.C.; Lam, C.S.; Pang, S.Y.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci. Rep. 2017, 18, 40887. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Yang, M.S.; Choi, D.; Kim, J.H.; Kim, H.S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.Y.; Joe, E.H. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS ONE 2012, 7, e34693. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Inoue, K.; Iwamura, H.; Terashima, K.; Soya, H.; Asashima, M.; Kuwabara, T. Reduction in paracrine Wnt3 factors during aging causes impaired adult neurogenesis. FASEB J. 2011, 25, 3570–3582. [Google Scholar] [CrossRef] [PubMed]
- Seib, D.R.M.; Corsini, N.S.; Ellwanger, K.; Plaas, C.; Mateos, A.; Pitzer, C.; Niehrs, C.; Celikel, T.; Martin-Villalba, A. Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell Stem Cell 2013, 12, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.J.; Braun, L.; Jiang, Y.; Hester, M.E.; Zhang, L.; Riolo, M.; Wang, H.; Rao, M.; Altura, R.A.; Kaspar, B.K. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signalling. Aging Cell 2012, 11, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.W.; McBryan, T.; Adams, P.D.; Sedivy, J.M. The effects of aging on the expression of Wnt pathway genes in mouse tissues. Age (Dordr) 2014, 36, 9618. [Google Scholar] [CrossRef] [PubMed]
- Orellana, A.M.M.; Vasconcelos, A.R.; Leite, J.A.; de Sá Lima, L.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-related neuroinflammation and changes in AKT-GSK-3β and WNT/β-CATENIN signaling in rat hippocampus. Aging 2015, 7, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Bayod, S.; Felice, P.; Andrés, P.; Rosa, P.; Camins, A.; Pallàs, M.; Canudas, A.M. Downregulation of canonical Wnt signaling in hippocampus of SAMP8 mice. Neurobiol. Aging 2015, 36, 720–729. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of synapse degeneration by restoring wnt signaling in the adult hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The secreted Wnt antagonist dickkopf-1 is required for amyloid β-mediated synaptic loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Alvarez-Buylla, A. Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 7526–7531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Builla, A.; Garcia-Verdugo, J.M.; Tramontin, A.D. A unified hypothesis on the lineage of neural stem cells. Nat. Rev. Neurosci. 2001, 2, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Stevens, C.F.; Gage, F.H. Astroglia induce neurogenesis from adult neural stem cells. Nature 2002, 417, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Lie, D.C.; Colamarino, S.A.; Song, H.G.; Désiré, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Diarie, A.R.; et al. Wnt signaling regulates adult hippocampal neurogenesis. Nature 2005, 473, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, G.; Sousa, K.M.; Bryja, V.; Pinto, L.; Wagner, J.; Arenas, E. Ventral midbrain glia express region-specific transcription factors and regulate dopaminergic neurogenesis through Wnt-5a secretion. Mol. Cell Neurosci. 2006, 31, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, G.; Rawal, N.; Arenas, E. GSK-3β inhibition/β-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J. Cell Sci. 2004, 117, 5731–5737. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Maisel, M.; Wegner, F.; Liebau, S.; Kim, D.W.; Gerlach, M.; Schwarz, J.; Kim, K.S.; Storch, A. Multipotent neural stem cells from the adult tegmentum with dopaminergic potential develop essential properties of functional neurons. Stem Cells 2006, 24, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Suess, C.; Fauser, M.; Kanzler, S.; Witt, M.; Fabel, K.; Schwarz, J.; Höglinger, G.U.; Storch, A. Rostro-caudal loss of cellular diversity within the periventricular regions of the ventricular system. Stem Cells 2009, 27, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Ooto, S.; Akagi, T.; Mandai, M.; Akaike, A.; Takahashi, M. Wnt signalling promotes regeneration in the retina of adult mammals. J. Neurosci. 2007, 27, 4210–4219. [Google Scholar] [CrossRef] [PubMed]
- Briona, L.K.; Poulain, F.E.; Mosimann, C.; Dorsky, R.I. Wnt/ß-catenin signaling is required for radial glial neurogenesis following spinal cord injury. Dev. Biol. 2015, 403, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.L.; Udeh, A.; Kalahasty, K.; Hackman, A.S. A growing field: The regulation of axonal regeneration by Wnt signaling. Neural. Regen. Res. 2018, 13, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Maretto, S.; Cordenonsi, M.; Dupont, S.; Braghetta, P.; Broccoli, V.; Hassan, A.B.; Volpin, D.; Bressan, G.M.; Piccolo, S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 3299–3304. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Storch, A. Endogenous regeneration in Parkinson’s disease: Do we need orthotopic dopaminergic neurogenesis? Stem Cells 2008, 26, 2749–2752. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.K.; Maisel, M.; Hermann, A.; Stirl, K.; Storch, A. Restorative approaches in Parkinson’s Disease: Which cell type wins the race? J. Neurol. Sci. 2010, 289, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Lie, D.C.; Dziewczapolski, G.; Willhoite, A.R.; Kaspar, B.K.; Shults, C.W.; Gage, F.H. The adult substantia nigra contains progenitor cells with neurogenic potential. J. Neurosci. 2002, 22, 6639–6649. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.Q.; Chen, Z.C.; Zhang, P.; Huang, H.J.; Wang, T.T.; Ding, Y.Q.; Qi, S.S.; Zhang, C.; Chen, S.X.; Zhou, P.; et al. Newborn dopaminergic neurons are associated with the migration and differentiation of SVZ-derived neural progenitors in a 6-OHDA-injected mouse model. Neuroscience 2017, 352, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Silva-Alvarez, C.; Arrazola, M.S.; Godoy, J.A.; Ordenes, D.; Inrsytosa, N.C. Canonical Wnt signalling protects hippocampal neurons from Aβ oligomers: Role of non-canonicalWnt-5a/Ca(2+) in mitochondrial dynamics. Front. Cell Neurosci. 2013, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Han, X.R.; Wen, X.; Wang, S.; Fan, S.H.; Zhuang, J.; Wang, Y.J.; Zhang, Z.F.; Li, M.Q.; Hu, B.; et al. Salidroside Protection Against Oxidative Stress Injury Through the Wnt/β-Catenin Signaling Pathway in Rats with Parkinson’s Disease. Cell Physiol. Biochem. 2018, 46, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Wang, Y.S.; Yuan, Y.J.; Wan, Z.H.; Yao, T.C.; Li, H.H.; Tang, P.F.; Mei, X.F. Neuroprotective effect of rapamycin on spinal cord injury via activation of the Wnt/β-catenin signaling pathway. Neural. Regen. Res. 2015, 10, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chong, Z.Z.; Shang, Y.C.; Maiese, K. Wnt1 inducible signaling pathway protein 1 (WISP1) blocks neurodegeneration through phosphoinositide 3 kinase/Akt1 and apoptotic mitochondrial signaling involving Bad, Bax, Bim, and Bcl-xL. Curr. Neurovascular Res. 2012, 9, 20–31. [Google Scholar] [CrossRef]
- Maiese, K. Novel applications of trophic factors, Wnt and WISP for neuronal repair and regeneration in metabolic disease. Neural. Regen. Res. 2015, 10, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. WISP1: Clinical insights for a proliferative and restorative member of the CCN family. Curr. Neurovasc. Res. 2014, 11, 378–389. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Serra, P.A.; Impagnatiello, F.; Morale, M.C.; Marchetti, B. Combining nitric oxide release with anti-inflammatory activity preserves nigrostriatal dopaminergic innervation and prevents motor impairment in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J. Neuroinflam. 2010, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Impagnatiello, F.; Marchetti, B. Switching the microglial harmful phenotype promotes lifelong restoration of subtantia nigra dopaminergic neurons from inflammatory neurodegeneration in aged mice. Rejuvenat. Res. 2011, 14, 411–424. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchetti, B. Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3743. https://doi.org/10.3390/ijms19123743
Marchetti B. Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2018; 19(12):3743. https://doi.org/10.3390/ijms19123743
Chicago/Turabian StyleMarchetti, Bianca. 2018. "Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 19, no. 12: 3743. https://doi.org/10.3390/ijms19123743
APA StyleMarchetti, B. (2018). Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences, 19(12), 3743. https://doi.org/10.3390/ijms19123743