Regulation of iNOS on Immune Cells and Its Role in Diseases


{kind=link}
Abstract
:1. Introduction
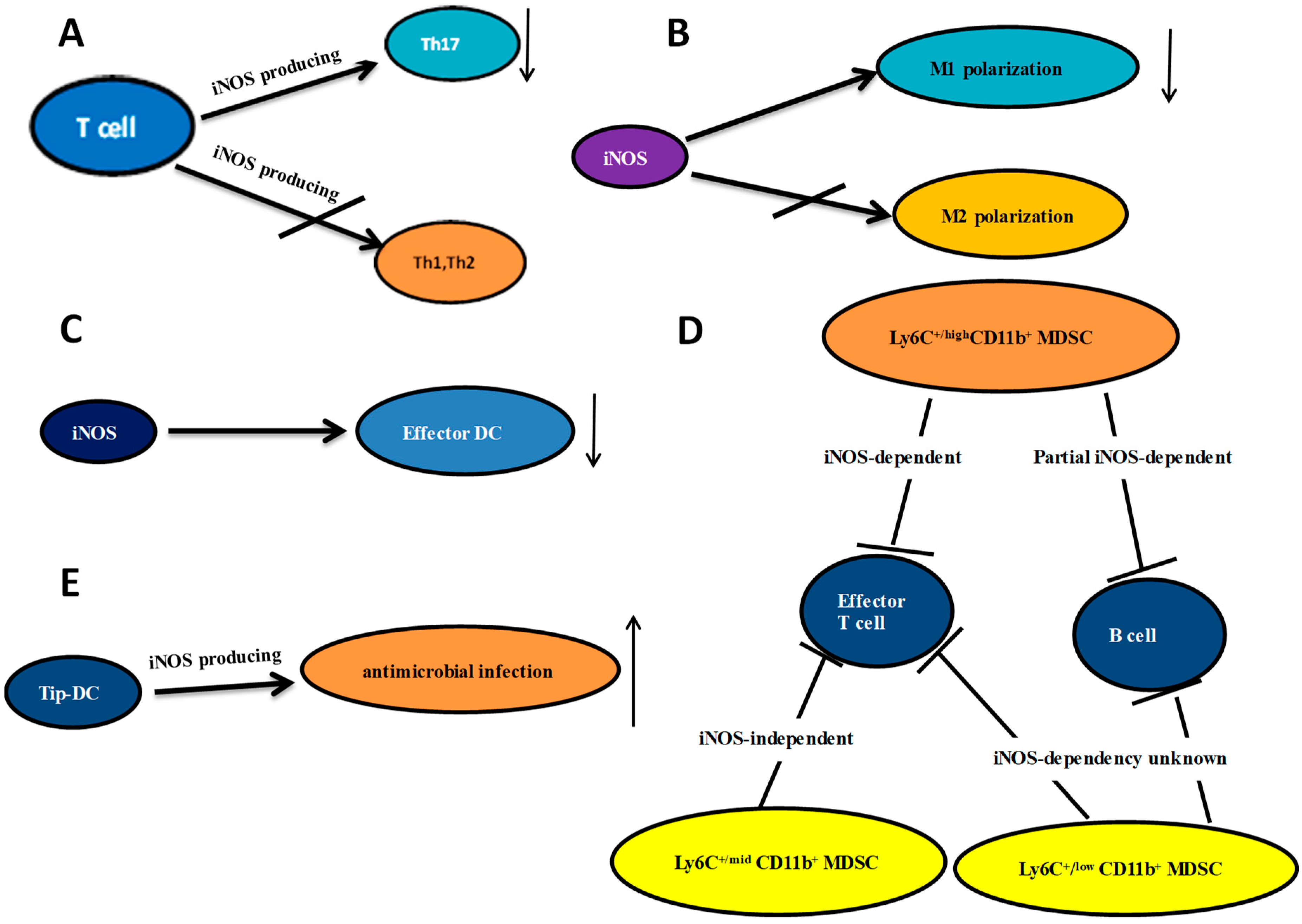
2. iNOS and T Cell Differentiation
3. The Effect of iNOS on Macrophages
4. Regulation of Dendritic Cells Differentiation by iNOS
5. Important Role of iNOS in MDSCs and Tip-DCs
6. The Role of iNOS in Cancer and Cancer Immunotherapy
7. Conclusions and Future Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.M.; Zhu, J. An introduction to nitric oxide sensing and response in bacteria. Adv. Appl. Microbiol. 2014, 87, 187–220. [Google Scholar] [PubMed]
- Carey, R.M.; Chen, B.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; Lee, R.J.; Cohen, N.A. Human upper airway epithelium produces nitric oxide in response to staphylococcus epidermidis. Int. Forum Allergy Rhinol. 2016, 6, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, V.A.; Barani, I.J.; Rabender, C.S.; Black, S.M.; Leach, J.K.; Graves, P.R.; Kellogg, G.E.; Mikkelsen, R.B. Tyrosine nitration of Ikappa Balpha: A novel mechanism for NF-kappaB activation. Biochemistry 2007, 46, 11671–11683. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, B.B.; Rathinam, V.A.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Niedbala, W.; Alves-Filho, J.C.; Fukada, S.Y.; Vieira, S.M.; Mitani, A.; Sonego, F.; Mirchandani, A.; Nascimento, D.C.; Cunha, F.Q.; Liew, F.Y. Regulation of type 17 helper T-cell function by nitric oxide during inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 9220–9225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Kawamura, I.; Nishibori, T.; Mitsuyama, M. Suppression of IFN-gamma production from Listeria monocytogenes-specifc T cells by endogenously produced nitric oxide. Cell. Immunol. 1996, 172, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Zhu, C.; Li, F.; Hegazi, R.; He, K.; Babyatsky, M.; Bauer, A.J.; Plevy, S.E. Inhibition of interleukin-12 p40 transcription and NFkappaB activation by nitric oxide in murine macrophages and dendritic cells. J. Biol. Chem. 2004, 279, 10776–10783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, Q.; Chuang, P.Y.; Liu, R.; Yang, J.; Peng, L.; Dai, Y.; Zheng, Z.; Qi, C.F.; He, J.C.; et al. Regulation of pathogenic Th17 cell differentiation by IL-10 in the development of glomerulonephritis. Am. J. Pathol. 2013, 183, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, R.; Lu, G.; Shen, Y.; Peng, L.; Zhu, C.; Cui, M.; Wang, W.; Arnaboldi, P.; Tang, M.; et al. T cell-derived inducible nitric oxide synthase swiches off TH17 cell differentiation. J. Exp. Med. 2013, 210, 1447–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermajer, N.; Wong, J.L.; Edwards, R.P.; Chen, K.; Scott, M.; Khader, S.; Kolls, J.K.; Odunsi, K.; Billiar, T.R.; Kalinski, P. Induction and stabiligy of human Th17 cells require endogenous NOS2 and cGMP-dependent NO signaling. J. Exp. Med. 2013, 210, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Niedbala, W.; Cai, B.; Liew, F.Y. Role of nitric oxide in the regulation of T cell functions. Ann. Rheum. Dis. 2006, 65, iii37–iii40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, C.; Röllinghoff, M.; Diefenbach, A. The role of nitric oxide in innate immunity. Immunol. Rev. 2000, 173, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Ze, H.; Chen, J.; Khan, F.U.; Chen, X.; Xu, J.; Ding, Q. The protective effects of a novel synthetic β-elemene derivative on human umbilical vein endothelial cells against oxidative stress-induced injury:involvement of antioxidation and PI3k/Akt/eNOS/NO signaling pathways. Biomed. Pharmacother. 2018, 106, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.P.; Niedbala, W.; Wei, X.Q.; Xu, D.; Feng, G.J.; Robinson, J.H.; Lam, C.; Liew, F.Y. Nitric oxide regulates Th1 cell development through the inhibition of IL-12 synthesis by macrophages. Eur. J. Immunol. 1998, 28, 4062–4070. [Google Scholar] [CrossRef] [Green Version]
- Nath, N.; Morinaga, O.; Singh, I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2010, 5, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Neverova, I.; Van Eyk, J.E.; Bennett, B.M. Nitration of tyrosine 92 mediates the activation of rat microsomal glutathione s-transferase by peroxynitrite. J. Biol. Chem. 2006, 281, 1986–1991. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Giri, S.; Nath, N.; Singh, I.; Singh, A.K. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007, 55, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.L.; Fang, H.X.; Liang, Y.; Zhao, Y.; Shi, C.S. MicroRNA-34a promotes iNOS secretion from pulmonary macrophages in septic suckling rats through activating STAT3 pathway. Biomed. Pharmacother. 2018, 105, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Jatana, M.; Elango, C.; Paintlia, A.S.; Singh, A.K.; Singh, I. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide 2006, 15, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.M.; Bishop, K.; Fox, D.; Zou, W. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011, 32, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitay, A.M.; Link, A.; Geibel, J.P. Activation of secretagogue independent gastric acid secretion via endothelial nitric oxide synthase stimulation in rats. Cell Physiol. Biochem. 2017, 44, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Gow, A. Pathophysiological functions of nitric oxidemediated protein modifications. Toxicology 2005, 208, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zhang, R.; Geng, S.; Peng, L.; Jayaraman, P.; Chen, C.; Xu, F.; Yang, J.; Li, Q.; Zheng, H.; et al. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat. Commun. 2015, 6, 6676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, D.; Li, C.; Suthar, M.S.; Draves, K.E.; Ma, D.Y.; Gale Jr, M.; Clark, E.A. Nitric oxide controls an inflammatory-like Ly6C(hi)PDCA1+DC subset that regulates Th1 immune responses. J. Leukoc. Biol. 2011, 89, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Suwanpradid, J.; Shih, M.; Pontius, L.; Yang, B.; Birukova, A.; Guttman-Yassky, E.; Corcoran, D.L.; Que, L.G.; Tighe, R.M.; MacLeod, A.S. Arginase 1 deficiency in monocytes/macrophages upregulates inducible nitric oxide synthase to promote cutaneous contact hypersensitivity. J Immunol. 2017, 199, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.S.; Sharman, S.K.; Lu, C.; Yang, D.; Paschall, A.V.; Tulachan, S.S.; Liu, K. The NF-κB p65 and p50 homodimer cooperate with IRF8 to activate iNOS transcription. BMC Cancer 2015, 15, 770. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Mladenov, R.; Huhn, M.; Barth, S.; Thepen, T. Macrophage targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases. Toxins 2012, 4, 676–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, H.B.; Mosser, D.M. Extrinsic and intrinsic control of macrophage inflammatory responses. J. Leukoc. Biol. 2013, 94, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, S.; Fitzsimons, A.; Boyd, C.R.; Kessler, J.; Fitzgerald, D.; Elliott, J.; Gabhann, J.N.; Smith, S.; Sica, A.; Hams, E.; et al. Suppressors of cytokine signaling 2 and 3 diametrically control macrophage polarization. Immunity 2013, 38, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ramos, T.; Carpio, Y.; Bolívar, J.; Gómez, L.; Estrada, M.P.; Pendón, C. Nitric oxide synthase-dependent immune response against gram negative bacteria in a crustacean, Litopenaeus vannamei. Fish Shellfish Immunol. 2016, 50, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.; Choi, H.; Eun, S.-Y.; Fukuyama, S.; Croft, M. Nitric oxide modulates TGF-beta-directive signals to suppress Foxp3+ regulatory T cell differentiation and potentiate Th1 development. J. Immunol. 2011, 186, 6972–6980. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Kolbeck, R.; Sanjuan, M.A. Plasmacytoid dendritic cells in autoimmunity. Curr. Opin. Immunol. 2017, 44, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Ambe, K.; Kon, H.; Takada, S.; Watanabe, H. Nitric oxide synthase (NOS) isoform expression after peripheral nerve transection in mice. Bull. Tokyo Dent. Coll. 2018, 59, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Si, C.; Zhang, R.; Wu, T.; Lu, G.; Hu, Y.; Zhang, H.; Xu, F.; Wei, P.; Chen, K.; Tang, H.; et al. Dendritic cell-derived nitric oxide inhibits the differentiation of effector dendritic cells. Oncotarget 2016, 7, 74834–74845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Tanabe, Y.; Ohya, S.; Mitsuyama, M. Administration of killed bacteria together with listeriolysin O induces protective immunity against Listeria monocytogenes in mice. Immunology 1998, 94, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Nino-Castro, A.C.; Schultze, J.L. Regulatory dendritic cells: There is more than just immune activation. Front. Immunol. 2012, 274, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 3, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Griffth, O.W.; Stueh, D. Nitric oxide synthases: Properties and catalytic mechanism. Annu. Rev. Physiol. 1995, 57, 707–736. [Google Scholar] [CrossRef] [PubMed]
- Zhong, K.; Song, W.; Wang, Q.; Wang, C.; Liu, X.; Chen, D.; Zhu, Z.; Wu, Y.; Zhang, W.; Zhang, M. Murine myeloid dendritic cells that phagocytose apoptotic T cells inhibit the immune response via NO. PLoS ONE 2012, 7, e49378. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, P.; Alfarano, M.G.; Svider, P.F.; Parikh, F.; Lu, G.; Kidwai, S.; Xiong, H.; Sikora, A.G. iNOS expression in CD4+ T cells limits Treg induction by repressing TGFβ1: Combined Inos inhibition and Treg depletion unmask endogenous antitumor immunity. Clin. Cancer Res. 2014, 20, 6439–6451. [Google Scholar] [CrossRef] [PubMed]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-derived suppressor cells express Bruton’s tyrosine kinase and can be depleted in tumor bearing hosts by ibrutinib treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Douguet, L.; Bod, L.; Lengagne, R.; Labarthe, L.; Kato, M.; Avril, M.F.; Prevost-Blondel, A. Nitric oxide synthase 2 is involved in the pro-tumorigenic potential of gammadelta17 T cells in melanoma. Oncoimmunology 2016, 5, e1208878. [Google Scholar] [CrossRef] [PubMed]
- James, S.L. Role of nitric oxide in parasitic infections. Microbiol. Rev. 1995, 59, 533–547. [Google Scholar] [PubMed]
- Burrack, K.S.; Morrison, T.E. The role of myeloid cell activation and arginine metabolism in the pathogenesis of virus-induced diseases. Front. Immunol. 2014, 5, 428. [Google Scholar] [CrossRef] [PubMed]
- Pereira, W.F.; Ribeiro-Gomes, F.L.; Guillermo, L.V.; Vellozo, N.S.; Montalvao, F.; Dosreis, G.A.; Lopes, M.F. Myeloid-derived suppressor cells help protective immunity to leishmania major infection despite suppressed T cell responses. J. Leukoc. Biol. 2011, 90, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Goni, O.; Alcaide, P.; Fresno, M. Immunosuppression during acute Trypanosoma cruzi infection: Involvement of Ly6G (Gr1(+))CD11b(+)immature myeloid suppressor cells. Int. Immunol. 2002, 14, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloidderived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [PubMed]
- Paschall, A.V.; Yang, D.; Lu, C.; Choi, J.H.; Li, X.; Liu, F.; Figueroa, M.; Oberlies, N.H.; Pearce, C.; Bollag, W.B.; et al. H3K9 trimethylation silences fas expression to confer colon carcinoma immune escape and 5-fluorouracil chemoresistance. J. Immunol. 2015, 195, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Sierra, RA.; Trillo, T.J.; Mohamed, E.; Yu, L.; Achyut, B.R.; Arbab, A.; Bradford, J.W.; Osborme, BA.; Miele, L.; Rodriguez, PC. Anti-jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res. 2017, 77, 5628–5638. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Salazar-Mather, T.P.; Biron, C.A.; Kuziel, W.A.; Pamer, E.G. TNF/iNOS producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 2003, 19, 59–70. [Google Scholar] [CrossRef]
- Virna, S.; Deckert, M.; Lutjen, S.; Soltek, S.; Foulds, K.E.; Shen, H.; Korner, H.; Sedgwick, J.D.; Schluter, D. TNF is important for pathogen control and limits brain damage in murine cerebral listeriosis. J. Immunol. 2006, 177, 3972–3982. [Google Scholar] [CrossRef] [PubMed]
- Bosschaerts, T.; Guilliams, M.; Stijlemans, B.; Morias, Y.; Engel, D.; Tacke, F.; Herin, M.; De Baetselier, P.; Beschin, A. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog. 2010, 6, e1001045. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Z.; Wong, K.L.; Lin, G.; Yang, C.M.; Wong, S.C.; Angeli, V.; Macary, P.A.; Kemeny, D.M. Human CD8 T cells drive Th1 responses through the differentiation of TNF/iNOS-producing dendritic cells. Eur. J. Immunol. 2011, 41, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, P.; Ritter, U.; Labbow, S.; Donhauser, N.; Rollinghoff, M.; Bogdan, C.; Korner, H. Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 2001, 166, 4012–4019. [Google Scholar] [CrossRef] [PubMed]
- Solodova, E.; Jablonska, J.; Weiss, S.; Lienenklaus, S. Production of IFN-beta during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS ONE 2011, 6, e18543. [Google Scholar] [CrossRef] [PubMed]
- Ritter, U.; Frischknecht, F.; Van, Z.G. Are neutrophils important host cells for Leishmania parasites? Trends Parasitol. 2009, 25, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.C.; Kar, W.T.; Fiona, H.S.W.; Yen, L.C.; Yafang, T.; Lai, G.N.; Veronique, A.; David, M.K. CD8 T cells regulate allergic contact dermatitis by modulating CCR2-dependent TNF/iNOS-expressing Ly6C+ CD11b+ monocytic cells. J. Investig. Dermatol. 2014, 134, 666–676. [Google Scholar]
- Grimm, EA.; Sikora, AG.; Ekmekcioglu, S. Molecular pathways: Inflammation-associated nitric-oxide production as a cancer-supporting redox mechanism and a potential therapeutic target. Clin Cancer Res. 2013, 19, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Ekmekcioglu, S.; Grimm, EA.; Roszik, J. Targeting iNOS to increase efficacy of immunotherapies. Hum. Vaccin. Immunother. 2017, 13, 1105–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmini, J.; Matthew, G.A.; Peter, F.S.; Falguni, P.; Geming, L.; Xiong, H.; Andrew, G.S. iNOS expression in CD4+ T cells limits T-reg induction by repressing TGFβ-1: Combined iNOS inhibition and T-reg depletion unmask endogenous anti-tumor immunity. Clin Cancer Res. 2014, 20, 6439–6451. [Google Scholar]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. https://doi.org/10.3390/ijms19123805
Xue Q, Yan Y, Zhang R, Xiong H. Regulation of iNOS on Immune Cells and Its Role in Diseases. International Journal of Molecular Sciences. 2018; 19(12):3805. https://doi.org/10.3390/ijms19123805
Chicago/Turabian StyleXue, Qingjie, Yingchun Yan, Ruihua Zhang, and Huabao Xiong. 2018. "Regulation of iNOS on Immune Cells and Its Role in Diseases" International Journal of Molecular Sciences 19, no. 12: 3805. https://doi.org/10.3390/ijms19123805
APA StyleXue, Q., Yan, Y., Zhang, R., & Xiong, H. (2018). Regulation of iNOS on Immune Cells and Its Role in Diseases. International Journal of Molecular Sciences, 19(12), 3805. https://doi.org/10.3390/ijms19123805