Comparison of Two Solid-Phase Extraction (SPE) Methods for the Identification and Quantification of Porcine Retinal Protein Markers by LC-MS/MS

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
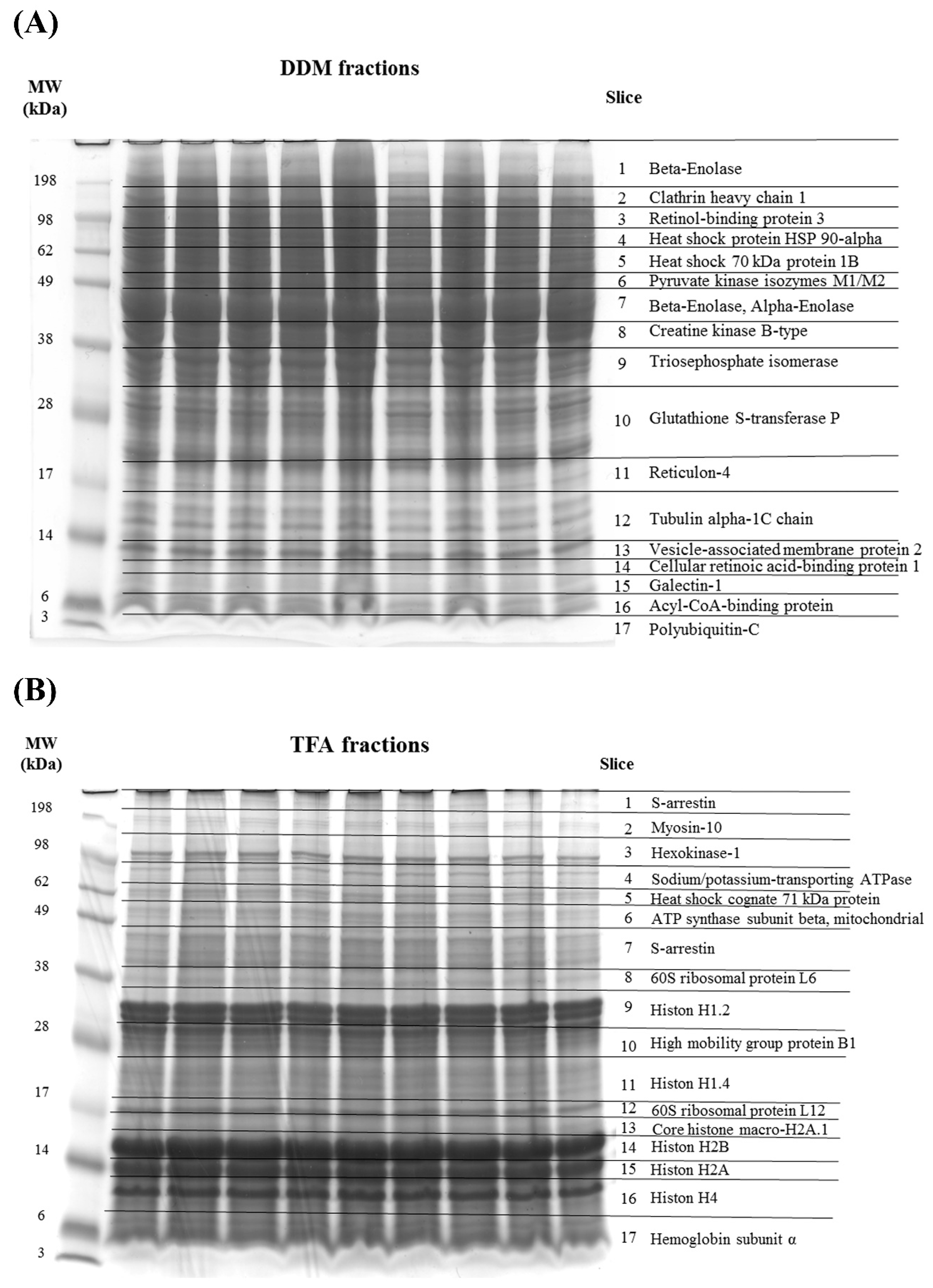
2.1. 1-D SDS Page of Porcine Retinal Proteins
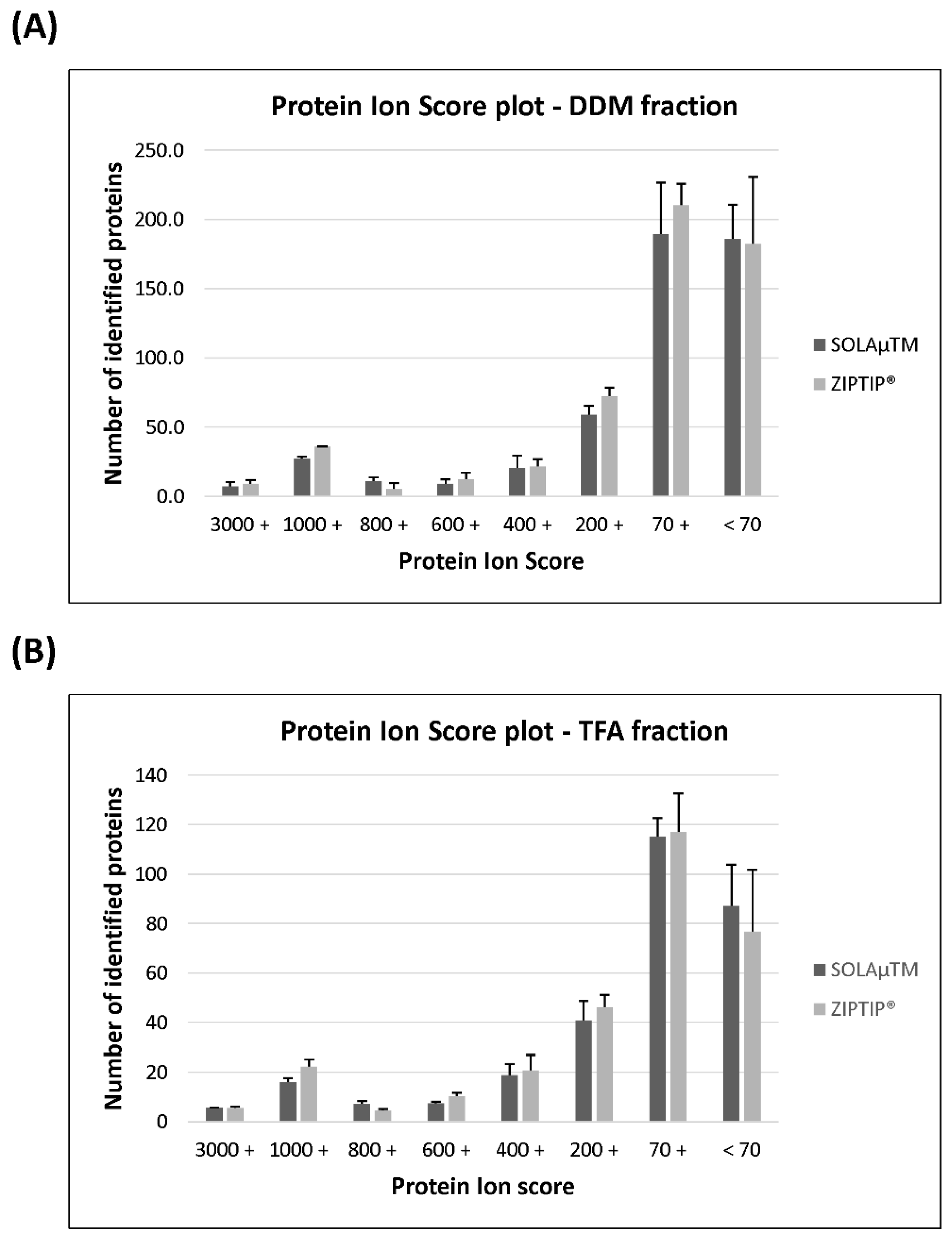
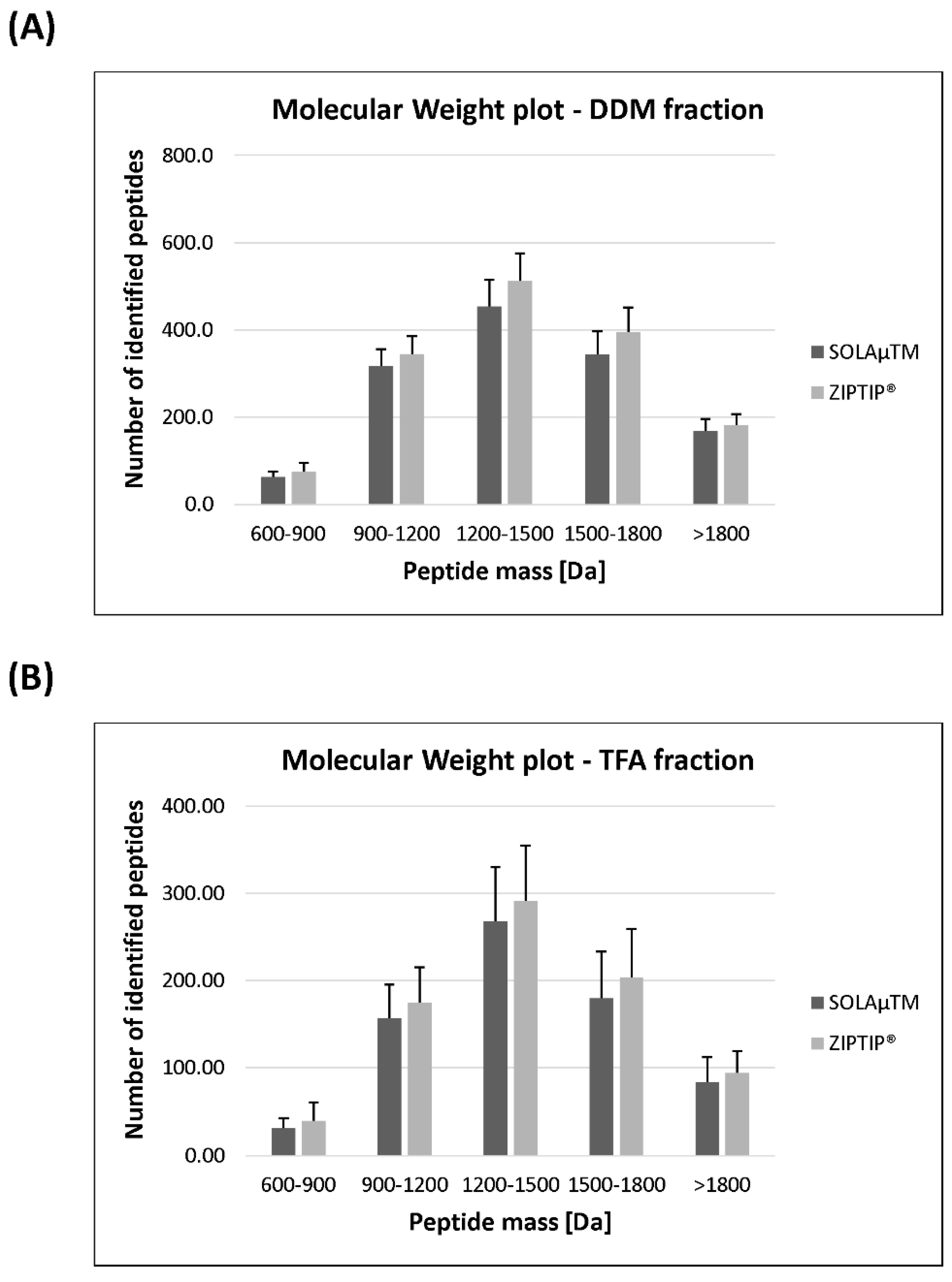
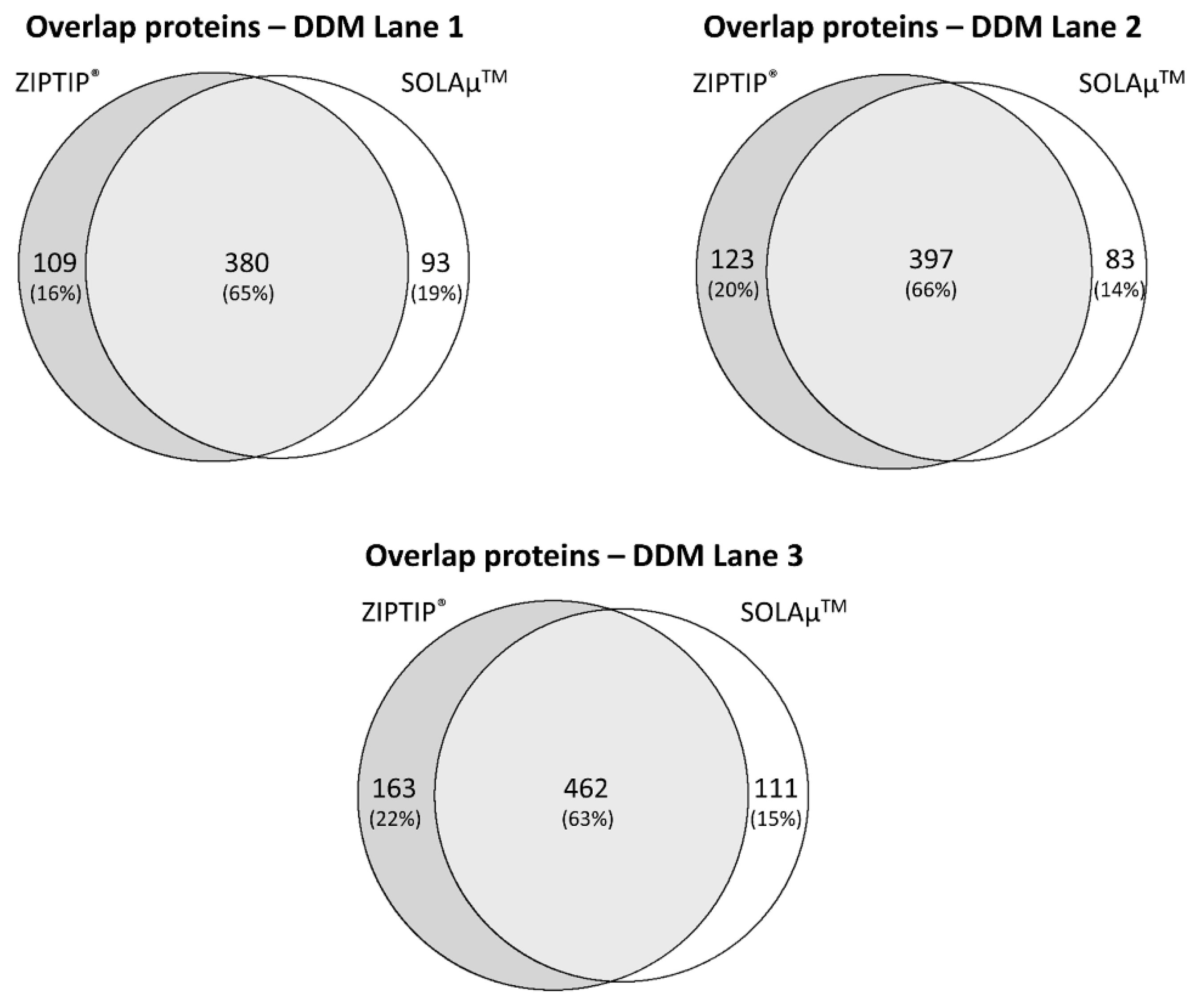
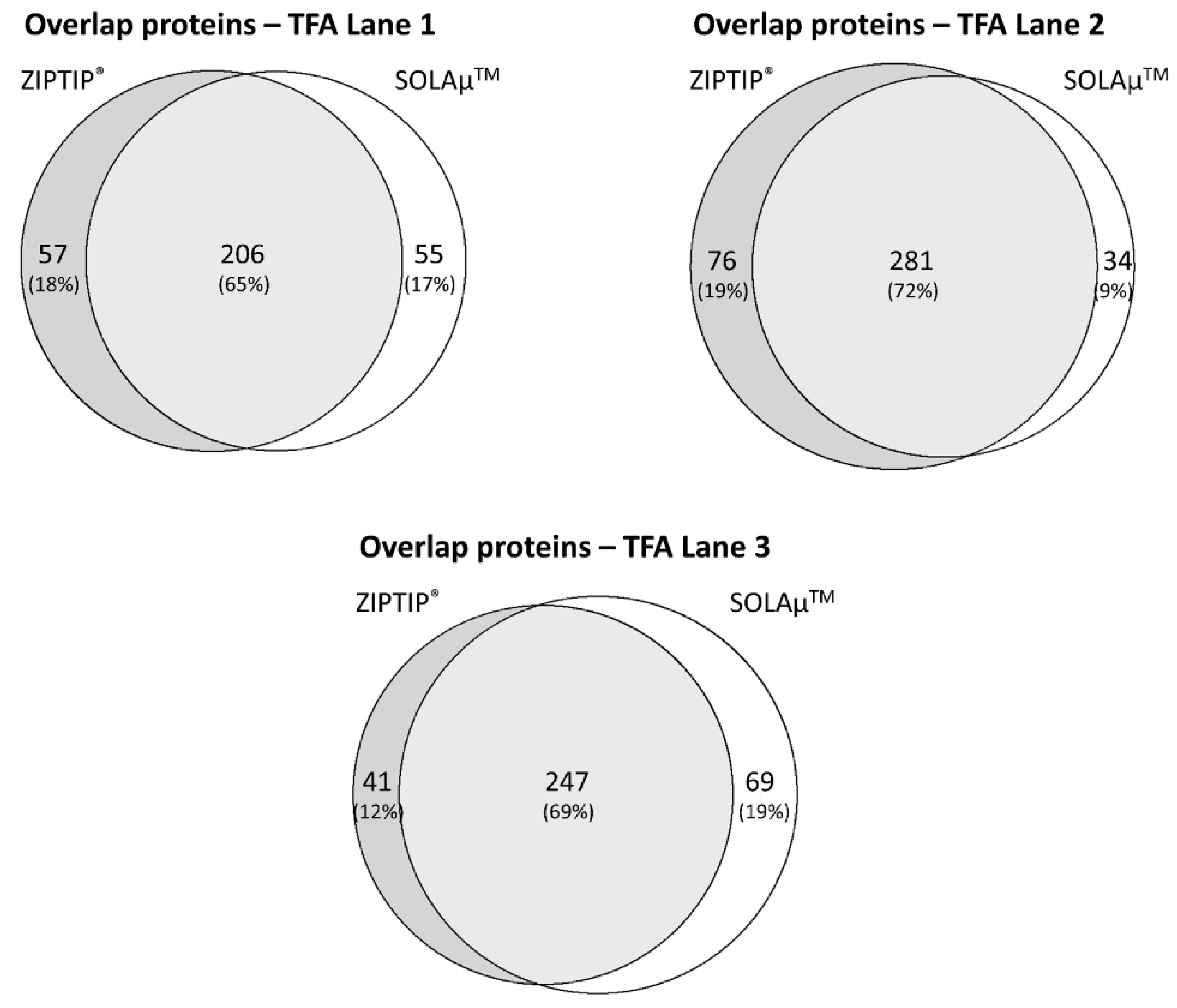
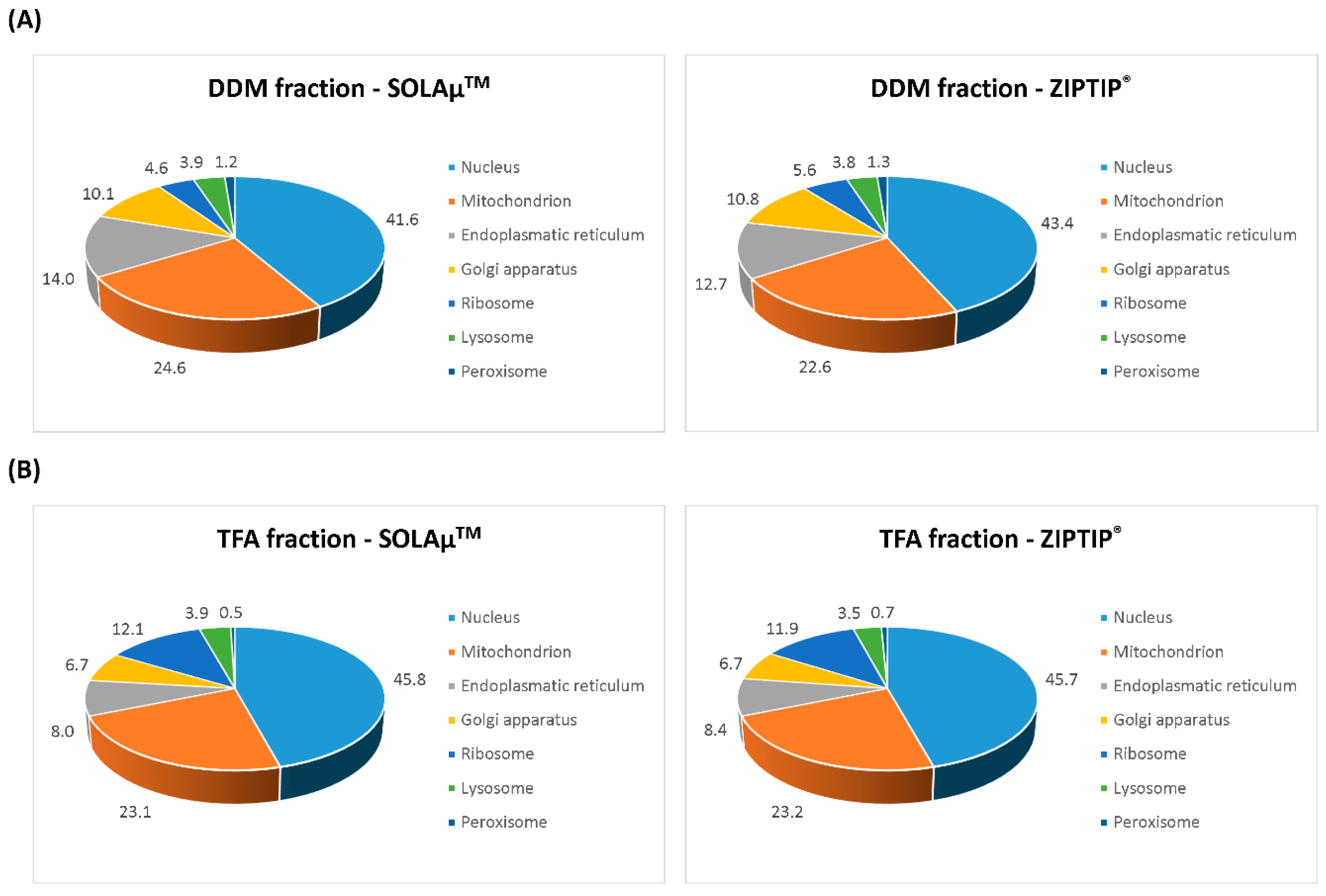
2.2. Qualitative Comparison between Both SPE-Based Peptide Purification Methods
2.3. Quantitative Analysis between Both SPE-Based Peptide Purification Methods
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and Protein Extraction Protocols
4.2. 1-D SDS Page
4.3. In-gel Trypsin Digestion
4.4. SPE-Based Peptide Purification
4.5. LC-MS/MS Analysis
4.6. Peptide Identification and Quantification
4.7. Data Analysis
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Hennion, M.C. Solid-phase extraction: Method development, sorbents, and coupling with liquid chromatography. J. Chromatogr. A 1999, 856, 3–54. [Google Scholar] [CrossRef]
- Poole, C.F. New trends in solid-phase extraction. TrAC Trends Anal. Chem. 2003, 22, 362–373. [Google Scholar] [CrossRef]
- Tamayo, F.G.; Turiel, E.; Martin-Esteban, A. Molecularly imprinted polymers for solid-phase extraction and solid-phase microextraction: Recent developments and future trends. J. Chromatogr. A 2007, 1152, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Augusto, F.; Hantao, L.W.; Mogollon, N.G.S.; Braga, S.C.G.N. New materials and trends in sorbents for solid-phase extraction. TrAC Trends Anal. Chem. 2013, 43, 14–23. [Google Scholar] [CrossRef]
- Bladergroen, M.R.; van der Burgt, Y.E.M. Solid-phase extraction strategies to surmount body fluid sample complexity in high-throughput mass spectrometry-based proteomics. J. Anal. Methods Chem. 2015, 2015, 250131. [Google Scholar] [CrossRef] [PubMed]
- Plotka-Wasylka, J.; Szczepanska, N.; de la Guardia, M.; Namiesnik, J. Modern trends in solid phase extraction: New sorbent media. TrAC Trends Anal. Chem. 2016, 77, 23–43. [Google Scholar] [CrossRef]
- Zwir-Ferenc, A.; Biziuk, M. Solid phase extraction technique—Trends, opportunities and applications. Pol. J. Environ. Stud. 2006, 15, 677–690. [Google Scholar]
- Funke, S.; Perumal, N.; Bell, K.; Pfeiffer, N.; Grus, F.H. The potential impact of recent insights into proteomic changes associated with glaucoma. Expert Rev. Proteom. 2017, 14, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Schmelter, C.; Perumal, N.; Funke, S.; Bell, K.; Pfeiffer, N.; Grus, F.H. Peptides of the variable igg domain as potential biomarker candidates in primary open-angle glaucoma (poag). Hum. Mol. Genet. 2017, 26, 4451–4464. [Google Scholar] [CrossRef] [PubMed]
- Perumal, N.; Funke, S.; Pfeiffer, N.; Grus, F.H. Characterization of lacrimal proline-rich protein 4 (prr4) in human tear proteome. Proteomics 2014, 14, 1698–1709. [Google Scholar] [CrossRef]
- Perumal, N.; Funke, S.; Wolters, D.; Pfeiffer, N.; Grus, F.H. Characterization of human reflex tear proteome reveals high expression of lacrimal proline-rich protein 4 (prr4). Proteomics 2015, 15, 3370–3381. [Google Scholar] [CrossRef] [PubMed]
- Tubaon, R.M.; Haddad, P.R.; Quirino, J.P. Sample clean-up strategies for ESI mass spectrometry applications in bottom-up proteomics: Trends from 2012 to 2016. Proteomics 2017, 17, 1700011. [Google Scholar] [CrossRef] [PubMed]
- Anders, F.; Teister, J.; Funke, S.; Pfeiffer, N.; Grus, F.; Thanos, S.; Prokosch, V. Proteomic profiling reveals crucial retinal protein alterations in the early phase of an experimental glaucoma model. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Anders, F.; Teister, J.; Liu, A.H.; Funke, S.; Grus, F.H.; Thanos, S.; von Pein, H.D.; Pfeiffer, N.; Prokosch, V. Intravitreal injection of ß-crystallin b2 improves retinal ganglion cell survival in an experimental animal model of glaucoma. PLoS ONE 2017, 12, e0175451. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.; Markowitsch, S.; Schmelter, C.; Perumal, N.; Mwiiri, F.K.; Gabel-Scheurich, S.; Pfeiffer, N.; Grus, F.H. In-depth proteomic analysis of the porcine retina by use of a four step differential extraction bottom up LC MS platform. Mol. Neurobiol. 2016, 54, 7262–7275. [Google Scholar] [CrossRef]
- Funke, S.; Perumal, N.; Beck, S.; Gabel-Scheurich, S.; Schmelter, C.; Teister, J.; Gerbig, C.; Gramlich, O.W.; Pfeiffer, N.; Grus, F.H. Glaucoma related proteomic alterations in human retina samples. Sci. Rep. 2016, 6, 29759. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Wilding, C.; Funke, S.; Perumal, N.; Beck, S.; Wolters, D.; Holz-Muller, J.; Pfeiffer, N.; Grus, F.H. Neuroprotective effects of antibodies on retinal ganglion cells in an adolescent retina organ culture. J. Neurochem. 2016, 139, 256–269. [Google Scholar] [CrossRef]
- McKay, G.J.; Campbell, L.; Oliver, M.; Brockbank, S.; Simpson, D.A.C.; Curry, W.J. Preparation of planar retinal specimens: Verification by histology, mrna profiling, and proteome analysis. Mol. Vis. 2004, 10, 240–247. [Google Scholar]
- Cehofski, L.J.; Kruse, A.; Kjaergaard, B.; Stensballe, A.; Honore, B.; Vorum, H. Dye-free porcine model of experimental branch retinal vein occlusion: A suitable approach for retinal proteomics. J. Ophthalmol. 2015, 2015, 839137. [Google Scholar]
- Hauck, S.M. Proteomic analysis of the porcine interphotoreceptor matrix. Proteomics 2005, 5, 4637. [Google Scholar] [CrossRef]
- Verma, N.; Rettenmeier, A.W.; Schmitz-Spanke, S. Recent advances in the use of sus scrofa (pig) as a model system for proteomic studies. Proteomics 2011, 11, 776–793. [Google Scholar] [CrossRef] [PubMed]
- Bassols, A.; Costa, C.; Eckersall, P.D.; Osada, J.; Sabria, J.; Tibau, J. The pig as an animal model for human pathologies: A proteomics perspective. Proteom. Clin. Appl. 2014, 8, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Bendixen, E.; Danielsen, M.; Larsen, K.; Bendixen, C. Advances in porcine genomics and proteomics-a toolbox for developing the pig as a model organism for molecular biomedical research. Brief Funct. Genom. 2010, 9, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Hesselager, M.O.; Codrea, M.C.; Sun, Z.; Deutsch, E.W.; Bennike, T.B.; Stensballe, A.; Bundgaard, L.; Moritz, R.L.; Bendixen, E. The pigpeptideatlas: A recource for systems biology in animal production and biomedicine. Proteomics 2016, 16, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L.; Vega-Montoto, L.; Rudnick, P.A.; Variyath, A.M.; Ham, A.J.; Bunk, D.M.; Kilpatrick, L.E.; Billheimer, D.D.; Blackman, R.K.; Cardasis, H.L.; et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2010, 9, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Parbel, A.; Pettersen, H.; Fenyo, D.; Bjorkesten, L. Reproducibility of LC-MS-Based protein identification. J. Exp. Bot. 2006, 57, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sadygov, R.G.; Yates, J.R., 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, N.; Lasaosa, M.; Tholey, A.; Heinzle, E.; van Dorsselaer, A.; Huber, C.G. Repeatability of peptide identifications in shotgun proteome analysis employing off-line two-dimensional chromatographic separations and ion-trap ms. J. Sep. Sci. 2009, 32, 1156–1164. [Google Scholar] [CrossRef]
- Awile, O.; Krisko, A.; Sbalzarini, I.F.; Zagrovic, B. Intrinsically disordered regions may lower the hydration free energy in proteins: A case study of nudix hydrolase in the bacterium deinococcus radiodurans. PLoS Comput. Biol. 2010, 6, e1000854. [Google Scholar] [CrossRef] [Green Version]
- Adams, V.H.; McBryant, S.J.; Wade, P.A.; Woodcock, C.L.; Hansen, J.C. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MECP2. J. Biol. Chem. 2007, 282, 15057–15064. [Google Scholar] [CrossRef]
- Hansen, J.C.; Wexler, B.B.; Rogers, D.J.; Hite, K.C.; Panchenko, T.; Ajith, S.; Black, B.E. DNA binding restricts the intrinsic conformational flexibility of methyl CPG binding protein 2 (mecp2). J. Biol. Chem. 2011, 286, 18938–18948. [Google Scholar] [CrossRef]
- Ausio, J.; de Paz, A.M.; Esteller, M. Mecp2: The long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 2014, 20, 487–498. [Google Scholar]
- Bardsley, J.; Jones, J.; Barratini, V.; Humphryes, P.C.; Liddicoat, T. Improvement in Speed and Reproducibility of Protein Digestion Utilizing Novel Sample Preparation Technology in a Full Solution Workflow; PN21209-EN 0515S; Thermo Fisher Scientific: Waltham, MA, USA, 2015. [Google Scholar]
- Harris, C.M. Finding the right robot for MALDI. Anal. Chem. 2001, 73, 447a–451a. [Google Scholar] [CrossRef] [PubMed]
- Scientific, T.F. Solaμ Spe Plates Technical Guide; Technical Guide, TG20947_E 04/14S; Thermo Fisher Scientific: Waltham, MA, USA, 2014. [Google Scholar]
- Namera, A.; Saito, T. Spin column extraction as a new sample preparation method in bioanalysis. Bioanalysis 2015, 7, 2171–2176. [Google Scholar] [CrossRef]
- Waldera-Lupa, D.M.; Etemad-Parishanzadeh, O.; Brocksieper, M.; Kirchgaessler, N.; Seidel, S.; Kowalski, T.; Montesinos-Rongen, M.; Deckert, M.; Schlegel, U.; Stuhler, K. Proteomic changes in cerebrospinal fluid from primary central nervous system lymphoma patients are associated with protein ectodomain shedding. Oncotarget 2017, 8, 110118–110132. [Google Scholar] [CrossRef] [PubMed]
- Sandvik, T.A.; Husa, A.; Buchmann, M.; Ludanes, E. Routine supercritical fluid chromatography tandem mass spectrometry method for determination of vitamin k1 extracted from serum with a 96-well solid-phase extraction method. J. Appl. Lab. Med. 2017, 1, jalm-2016. [Google Scholar] [CrossRef]
- Rosmark, O.; Ahrman, E.; Muller, C.; Elowsson Rendin, L.; Eriksson, L.; Malmstrom, A.; Hallgren, O.; Larsson-Callerfelt, A.K.; Westergren-Thorsson, G.; Malmstrom, J. Quantifying extracellular matrix turnover in human lung scaffold cultures. Sci. Rep. 2018, 8, 5409. [Google Scholar] [CrossRef] [PubMed]
- Grassetti, A.V.; Hards, R.; Gerber, S.A. Offline pentafluorophenyl (PFP)-rp prefractionation as an alternative to high-PH RP for comprehensive LC-MS/MS proteomics and phosphoproteomics. Anal. Bioanal. Chem. 2017, 409, 4615–4625. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protocols 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the pride database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef]
- Dawson, H.D.; Chen, C.; Gaynor, B.; Shao, J.; Urban, J.F., Jr. The porcine translational research database: A manually curated, genomics and proteomics-based research resource. BMC Genom. 2017, 18, 643. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.M.; Bendixen, E. Pig proteomics: A review of a species in the crossroad between biomedical and food sciences. J. Proteom. 2012, 75, 4296–4314. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. 1d and 2d annotation enrichment: A statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 2012, 13 (Suppl. 16), S12. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmelter, C.; Funke, S.; Treml, J.; Beschnitt, A.; Perumal, N.; Manicam, C.; Pfeiffer, N.; Grus, F.H. Comparison of Two Solid-Phase Extraction (SPE) Methods for the Identification and Quantification of Porcine Retinal Protein Markers by LC-MS/MS. Int. J. Mol. Sci. 2018, 19, 3847. https://doi.org/10.3390/ijms19123847
Schmelter C, Funke S, Treml J, Beschnitt A, Perumal N, Manicam C, Pfeiffer N, Grus FH. Comparison of Two Solid-Phase Extraction (SPE) Methods for the Identification and Quantification of Porcine Retinal Protein Markers by LC-MS/MS. International Journal of Molecular Sciences. 2018; 19(12):3847. https://doi.org/10.3390/ijms19123847
Chicago/Turabian StyleSchmelter, Carsten, Sebastian Funke, Jana Treml, Anja Beschnitt, Natarajan Perumal, Caroline Manicam, Norbert Pfeiffer, and Franz H. Grus. 2018. "Comparison of Two Solid-Phase Extraction (SPE) Methods for the Identification and Quantification of Porcine Retinal Protein Markers by LC-MS/MS" International Journal of Molecular Sciences 19, no. 12: 3847. https://doi.org/10.3390/ijms19123847
APA StyleSchmelter, C., Funke, S., Treml, J., Beschnitt, A., Perumal, N., Manicam, C., Pfeiffer, N., & Grus, F. H. (2018). Comparison of Two Solid-Phase Extraction (SPE) Methods for the Identification and Quantification of Porcine Retinal Protein Markers by LC-MS/MS. International Journal of Molecular Sciences, 19(12), 3847. https://doi.org/10.3390/ijms19123847