Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy


Abstract
:
1. Introduction
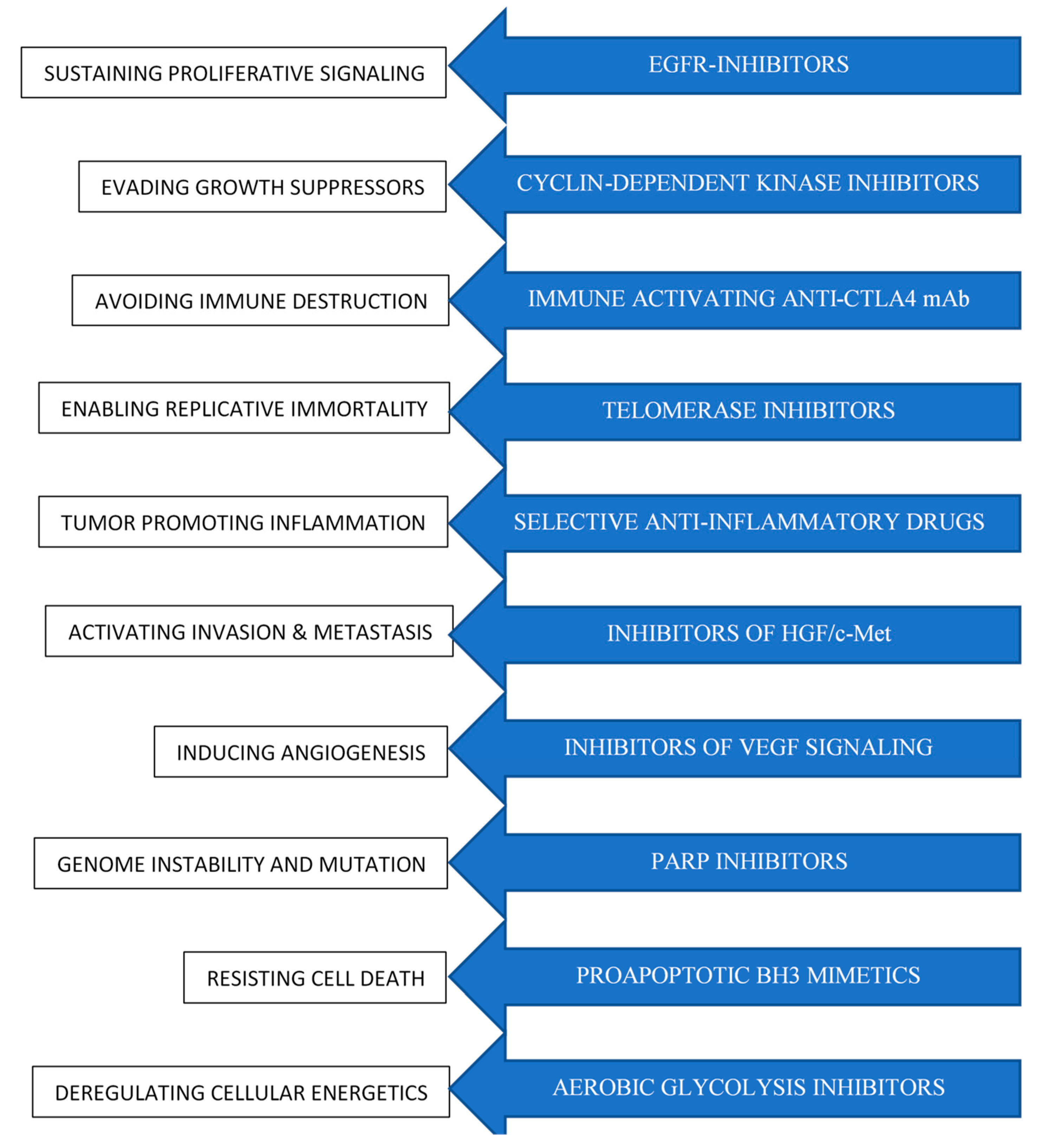
1.1. Current Overview of Cancer Therapeutics
1.2. Programmed Cell Death
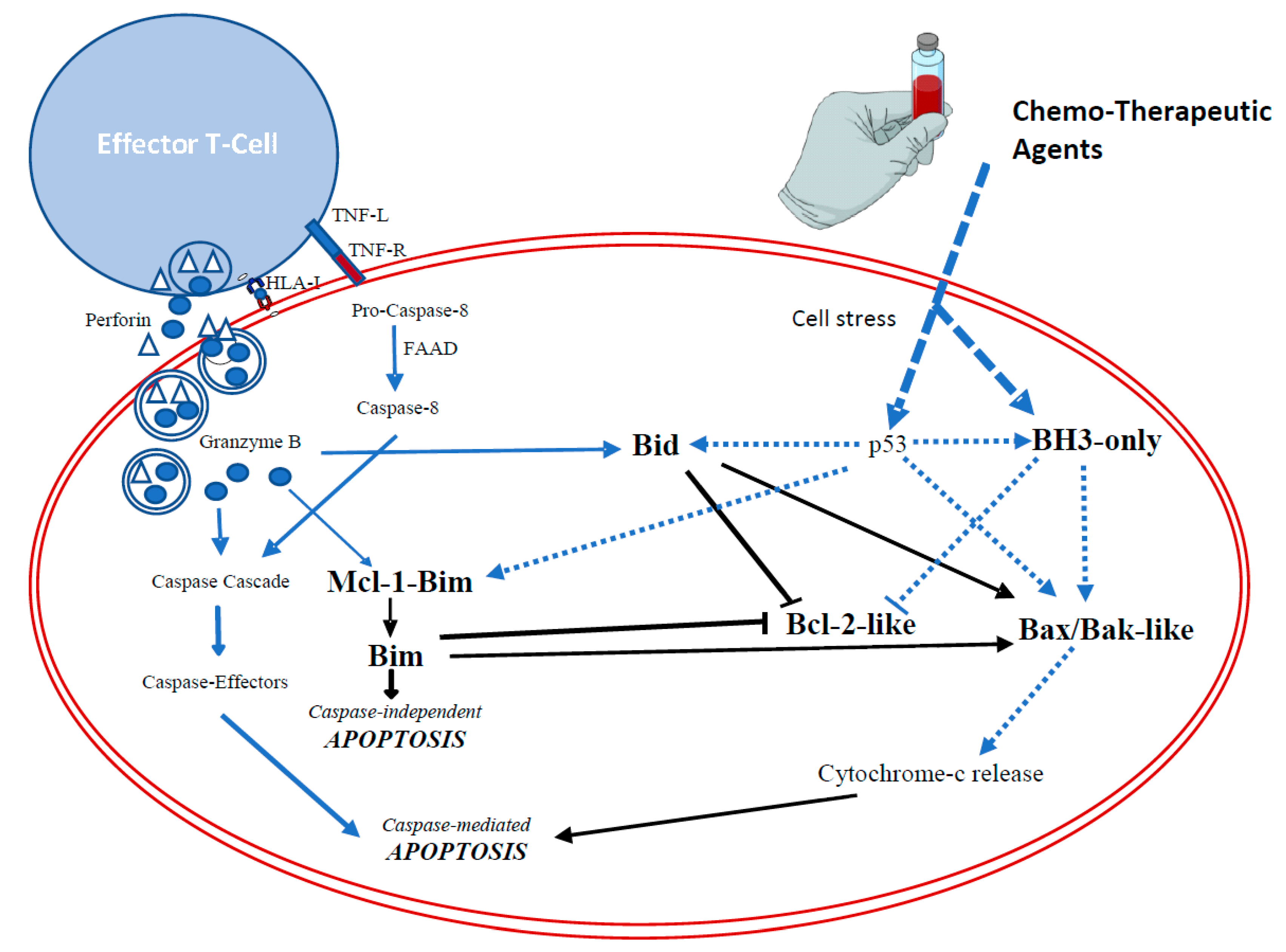
1.2.1. The Extrinsic Pathway
Death-receptor-mediated apoptosis
- TNFR1 (also known as death receptors or p55/p60 [21]): a death-domain-containing-protein in the intracellular portion which activates apoptosis via activation of intracellular death-inducing signaling complex (DISC) proteins [21], including Fas-associated proteins with death domain (FADD), TNFR1-associated death domain protein (TRADD) and other death domain-binding partners [20]. Most cells express constitutive but low levels of TNFR1 [21].
- Decoy receptors (DcR) with no intracellular interacting partners that act as TNF superfamily ligand inhibitors [20].
The cytotoxic granule-mediated cell death
1.2.2. The Intrinsic Pathway
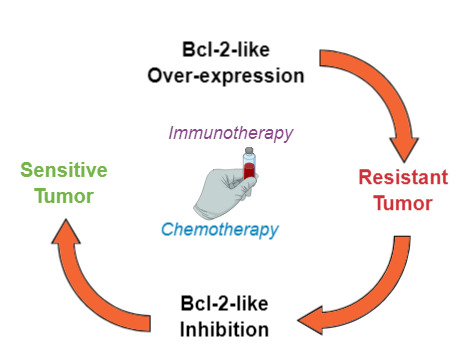
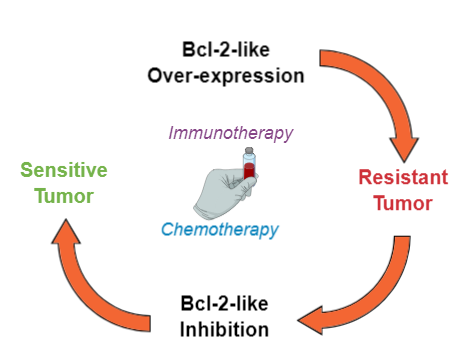
1.3. Resistance to Chemo- or Immunotherapy-Induced Apoptosis
2. BCL-2 as a Target for Cancer Treatment
2.1. The Bcl-2 Family
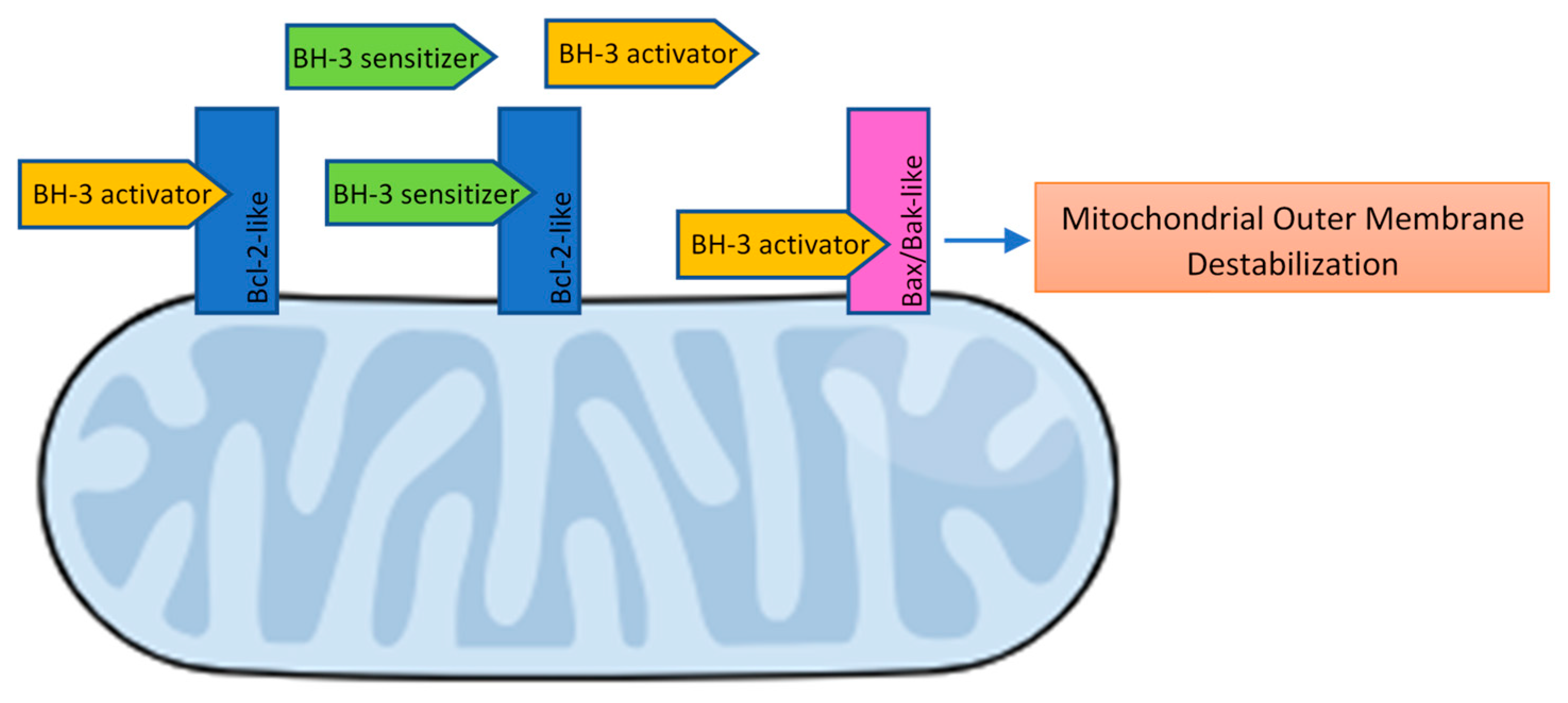
- Bcl-2 apoptosis effectors (Bax, Bak, Bok), whose oligomerization plays a central role during mitochondrial outer membrane destabilization, apoptogenic proteins (cytochrome c, SMAC, Htra2, etc.) release and caspase activation [89], are tightly controlled by other Bcl-2 proteins [18]. Recent studies also propose a “membrane-induced spontaneous Bax/Bak activation model” in which the outer mitochondrial membrane plays a central role in Bax/Bak activation regardless of the presence of other Bcl-2 proteins [90].
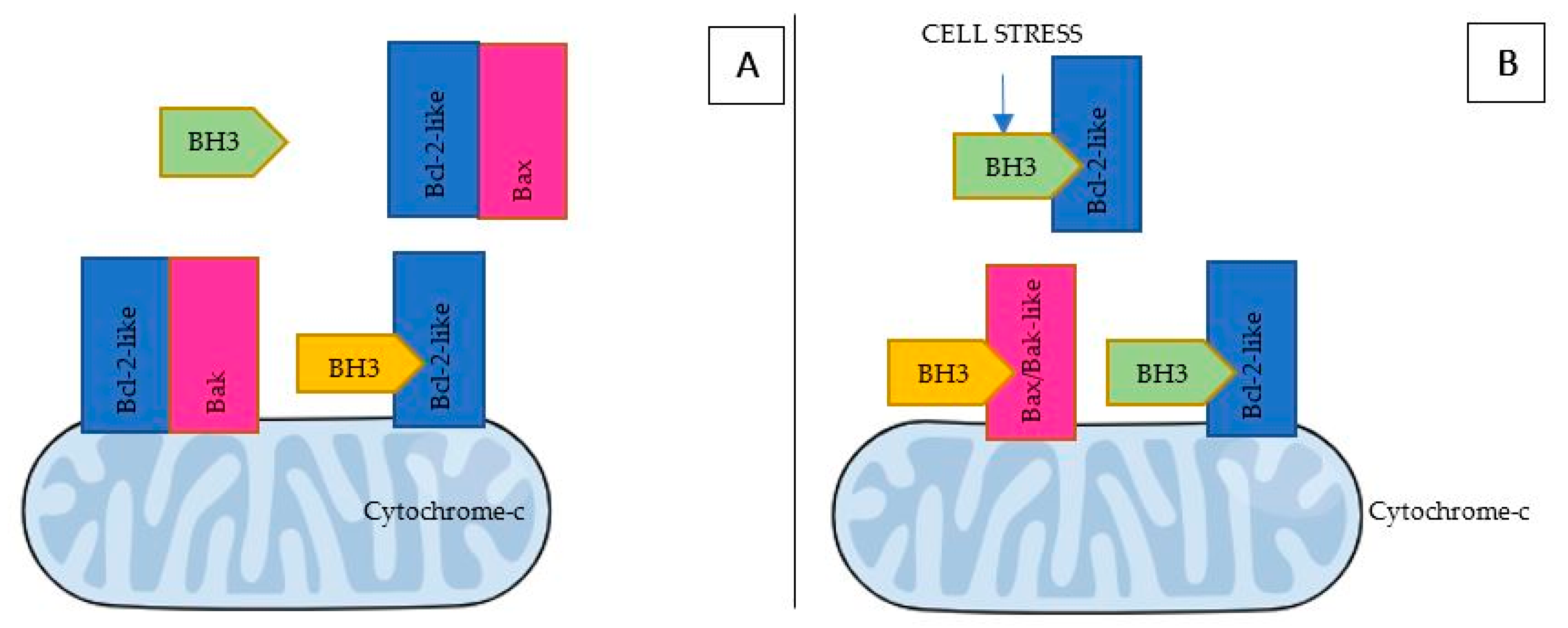
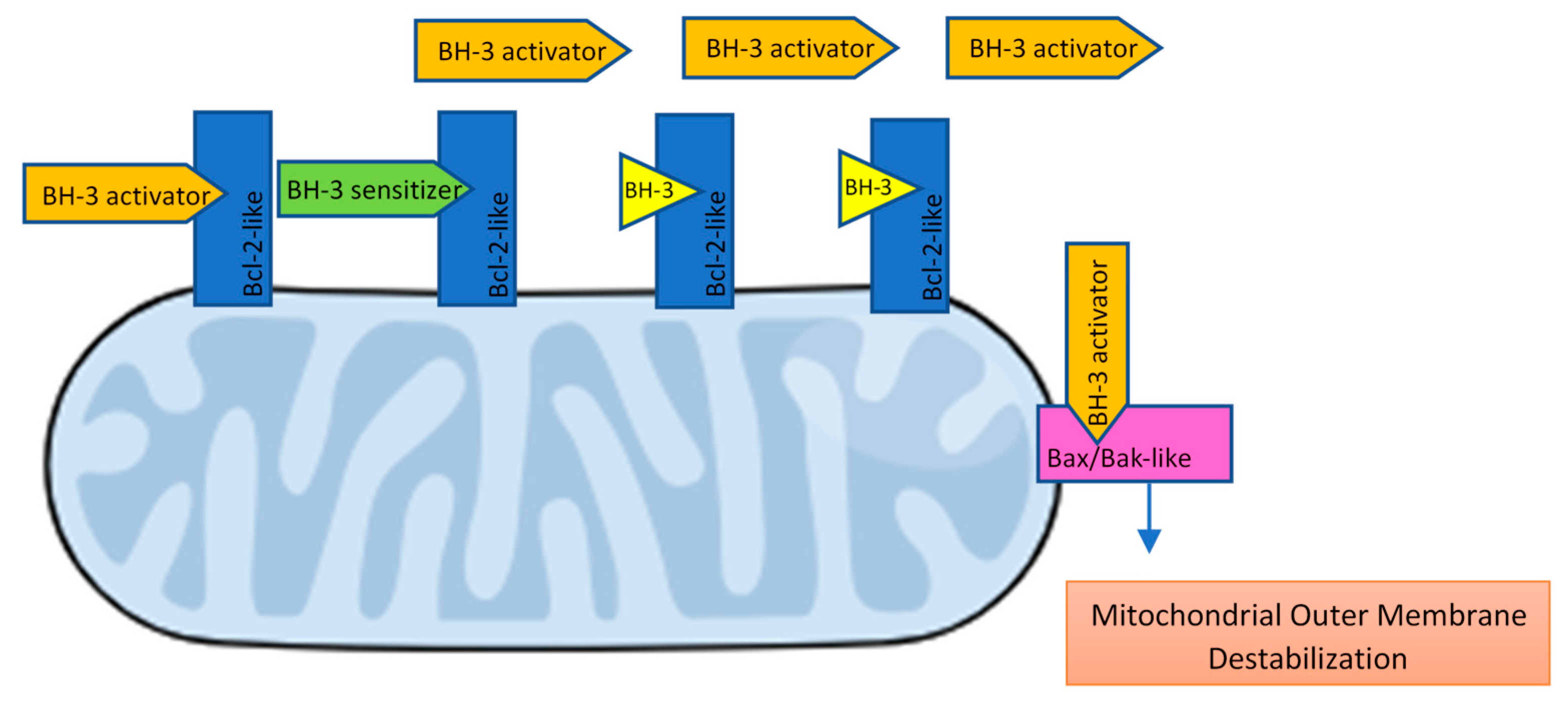
- BH3 (only) pro-apoptotic initiators [91] are able to respond to cellular stress by, directly or indirectly, activating the apoptosis effector (or executioner) members. These proteins are tightly regulated on the transcriptional and post-transcriptional level and can act as apoptosis sensitizers or activators [18]. BH3-activators (Bid, Bim, Puma, Bmf) bind to both the pro-apoptotic and the anti-apoptotic Bcl-2 multi-region members whilst BH3-sensitizers (Bad, Noxa, Hrk, Bik) unbind activator BH3-proteins and bind to the anti-apoptotic members, boosting mitochondrial outer membrane permeabilization [41,90]. Excess sensitizer proteins prevent activator-proteins sequestration by anti-apoptotic homologues, allowing them to directly interact with and activate the apoptosis effectors [67] (Figure 2). Both the amphipathic alpha-helical BH3-domain and the presence of the apoptotic effector members, are essential for BH3-only proteins death function [92].
- Bcl-2 anti-apoptotic members (Bcl-2-like proteins: Bcl-2, Bcl-xL, Bcl-W, Mcl-1, Bcl-B, Bcl2A1) [91], act directly preventing effectors oligomerization or indirectly sequestering and inactivating BH3-only proteins to prevent the effectors’ activation. Thus, the anti-apoptotic members can block both the BH3 (only) as well as the effector members [92] (Figure 3).
2.2. BCL-2 Inhibitors to Overcome Resistance to Anti-Cancer Treatments
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer Immunotherapy, Part 1: Current Strategies and Agents. Pharm. Ther. 2017, 42, 375–383. [Google Scholar]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buque, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. Cancer Immunotherapy, Part 2: Efficacy, Safety, and Other Clinical Considerations. Pharm. Ther. 2017, 42, 452–463. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. Biomed. Res. Int. 2014, 2014, 616149. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A., Jr.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: The Universal Protein Knowledgebase: Casp6_Human. Nucleic Acids Research 2018 Nov 11, 2018; 2699. Available online: https://www.uniprot.org/uniprot/P55212 (accessed on 11 November 2018).
- Jimenez Fernandez, D.; Lamkanfi, M. Inflammatory caspases: Key regulators of inflammation and cell death. Biol. Chem. 2015, 396, 193–203. [Google Scholar]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Rousalova, I.; Krepela, E. Granzyme B-induced apoptosis in cancer cells and its regulation (review). Int. J. Oncol. 2010, 37, 1361–1378. [Google Scholar]
- Vogler, M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef]
- Ware, C.F. The TNF superfamily. Cytokine Growth Factor Rev. 2003, 14, 181–184. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal 2018, 11. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J. Initiator caspases in apoptosis signaling pathways. Apoptosis 2002, 7, 313–319. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krähenbühl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef]
- Janssens, S.; Tinel, A. The PIDDosome, DNA-damage-induced apoptosis and beyond. Cell Death Differ. 2012, 19, 13–20. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef] [Green Version]
- Westphal, D.; Kluck, R.M.; Dewson, G. Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196–205. [Google Scholar] [CrossRef]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liu, H.; Xue, R.; Tang, W.; Zhang, S. BH3 Mimetic ABT-199 Enhances the Sensitivity of Gemcitabine in Pancreatic Cancer in vitro and in vivo. Dig. Dis. Sci. 2018, 63, 3367–3375. [Google Scholar] [CrossRef]
- Redondo, M.; Garcia, J.; Villar, E.; Rodrigo, I.; Perea-Milla, E.; Serrano, A.; Morell, M. Major histocompatibility complex status in breast carcinogenesis and relationship to apoptosis. Hum. Pathol. 2003, 34, 1283–1289. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. 1), 35–48. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Tupone, M.G.; Desideri, M.; Di Martile, M.; Gabellini, C.; Buglioni, S.; Pallocca, M.; Alessandrini, G.; D’Aguanno, S.; Del Bufalo, D. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 2017, 8, 3216. [Google Scholar] [CrossRef]
- Schenk, R.L.; Strasser, A.; Dewson, G. BCL-2: Long and winding path from discovery to therapeutic target. Biochem. Biophys. Res. Commun. 2017, 482, 459–469. [Google Scholar] [CrossRef]
- Nakamura, M.; Shimada, K.; Konishi, N. The role of HRK gene in human cancer. Oncogene 2008, 27 (Suppl. 1), S105–S113. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef]
- Greenberg, E.F.; Lavik, A.R.; Distelhorst, C.W. Bcl-2 regulation of the inositol 1,4,5-trisphosphate receptor and calcium signaling in normal and malignant lymphocytes: Potential new target for cancer treatment. Biochim. Biophys. Acta 2014, 1843, 2205–2210. [Google Scholar] [CrossRef]
- Frenzel, A.; Grespi, F.; Chmelewskij, W.; Villunger, A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis 2009, 14, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Ozretic, P.; Alvir, I.; Sarcevic, B.; Vujaskovic, Z.; Rendic-Miocevic, Z.; Roguljic, A.; Beketic-Oreskovic, L. Apoptosis regulator Bcl-2 is an independent prognostic marker for worse overall survival in triple-negative breast cancer patients. Int. J. Biol. Markers 2018, 33, 109–115. [Google Scholar] [CrossRef]
- Yang, D.; Chen, M.B.; Wang, L.Q.; Yang, L.; Liu, C.Y.; Lu, P.H. Bcl-2 expression predicts sensitivity to chemotherapy in breast cancer: A systematic review and meta-analysis. J. Exp. Clin. Cancer Res. 2013, 32, 105. [Google Scholar] [CrossRef]
- Villar, E.; Redondo, M.; Rodrigo, I.; Garcia, J.; Avila, E.; Matilla, A. bcl-2 Expression and apoptosis in primary and metastatic breast carcinomas. Tumour Biol. 2001, 22, 137–145. [Google Scholar] [CrossRef]
- Huang, Q.; Li, S.; Cheng, P.; Deng, M.; He, X.; Wang, Z.; Yang, C.-H.; Zhao, X.-Y.; Huang, J. High expression of anti-apoptotic protein Bcl-2 is a good prognostic factor in colorectal cancer: Result of a meta-analysis. World J. Gastroenterol. 2017, 23, 5018. [Google Scholar] [CrossRef]
- Itoi, T.; Yamana, K.; Bilim, V.; Takahashi, K.; Tomita, F. Impact of frequent Bcl-2 expression on better prognosis in renal cell carcinoma patients. Br. J. Cancer 2004, 90, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Hidaka, S.; Tagawa, Y.; Nagayasu, T. Bcl-2 protein expression correlates with better prognosis in patients with advanced non-small cell lung cancer. Anticancer Res. 2004, 24, 1925–1928. [Google Scholar]
- Chipuk, J.E.; Bouchier-Hayes, L.; Kuwana, T.; Newmeyer, D.D.; Green, D.R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005, 309, 1732–1735. [Google Scholar] [CrossRef]
- Kaufmann, T.; Schinzel, A.; Borner, C. Bcl-w(edding) with mitochondria. Trends Cell Biol. 2004, 14, 8–12. [Google Scholar] [CrossRef]
- Bae, I.H.; Park, M.J.; Yoon, S.H.; Kang, S.W.; Lee, S.S.; Choi, K.M.; Um, H.D. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res. 2006, 66, 4991–4995. [Google Scholar] [CrossRef]
- Wilson, J.W.; Nostro, M.C.; Balzi, M.; Faraoni, P.; Cianchi, F.; Becciolini, A.; Potten, C.S. Bcl-w expression in colorectal adenocarcinoma. Br. J. Cancer 2000, 82, 178–185. [Google Scholar] [CrossRef]
- Adams, C.M.; Kim, A.S.; Mitra, R.; Choi, J.K.; Gong, J.Z.; Eischen, C.M. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J. Clin. Investig. 2017, 127, 635–650. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.M.; Mitra, R.; Gong, J.; Eischen, C.M. Non-Hodgkin and Hodgkin lymphomas select for overexpression of BCLW. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Garofalo, M.; Quintavalle, C.; Zanca, C.; De Rienzo, A.; Romano, G.; Acunzo, M.; Puca, L.; Incoronato, M.; Croce, C.M.; Condorelli, G. Akt regulates drug-induced cell death through Bcl-w downregulation. PLoS ONE 2008, 3, e4070. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Hind, C.K.; Carter, M.J.; Harris, C.L.; Chan, H.T.; James, S.; Cragg, M.S. Role of the pro-survival molecule Bfl-1 in melanoma. Int. J. Biochem. Cell Biol. 2015, 59, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Ke, N.; Godzik, A.; Reed, J.C. Bcl-B, a novel Bcl-2 family member that differentially binds and regulates Bax and Bak. J. Biol. Chem. 2001, 276, 12481–12484. [Google Scholar] [CrossRef]
- Hamouda, M.-A.; Jacquel, A.; Robert, G.; Puissant, A.; Richez, V.; Cassel, R.; Fenouille, N.; Roulland, S.; Gilleron, J.; Griessinger, E. BCL-B (BCL2L10) is overexpressed in patients suffering from multiple myeloma (MM) and drives an MM-like disease in transgenic mice. J. Exp. Med. 2016, 213, 1705–1722. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Llambi, F.; Wang, Y.M.; Victor, B.; Yang, M.; Schneider, D.M.; Gingras, S.; Parsons, M.J.; Zheng, J.H.; Brown, S.A.; Pelletier, S.; et al. BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell 2016, 165, 421–433. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Di Giovanni, S.; Wang, G.; Liu, W.; Stoica, B.; Faden, A.I. BOK and NOXA are essential mediators of p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 28367–28374. [Google Scholar] [CrossRef]
- Sax, J.K.; Fei, P.; Murphy, M.E.; Bernhard, E.; Korsmeyer, S.J.; El-Deiry, W.S. BID regulation by p53 contributes to chemosensitivity. Nat. Cell Biol. 2002, 4, 842–849. [Google Scholar] [CrossRef]
- Song, G.; Wang, W.; Hu, T. p53 facilitates BH3-only BID nuclear export to induce apoptosis in the irrepairable DNA damage response. Med. Hypotheses 2011, 77, 850–852. [Google Scholar] [CrossRef]
- Grespi, F.; Soratroi, C.; Krumschnabel, G.; Sohm, B.; Ploner, C.; Geley, S.; Hengst, L.; Häcker, G.; Villunger, A. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010, 17, 1672. [Google Scholar] [CrossRef]
- Esposti, M.D. The roles of Bid. Apoptosis 2002, 7, 433–440. [Google Scholar] [CrossRef]
- Lee, J.H.; Soung, Y.H.; Lee, J.W.; Park, W.S.; Kim, S.Y.; Cho, Y.G.; Kim, C.J.; Seo, S.H.; Kim, H.S.; Nam, S.W. Inactivating mutation of the pro-apoptotic gene BID in gastric cancer. J. Pathol. J. Pathol. Soc. G. B. Irel. 2004, 202, 439–445. [Google Scholar]
- Goncharenko-Khaider, N.; Lane, D.; Matte, I.; Rancourt, C.; Piche, A. The inhibition of Bid expression by Akt leads to resistance to TRAIL-induced apoptosis in ovarian cancer cells. Oncogene 2010, 29, 5523–5536. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Goldstein, L.A.; Hou, W.; Gastman, B.R.; Rabinowich, H. Regulation of mitochondrial apoptotic events by p53-mediated disruption of complexes between anti-apoptotic Bcl-2 members and Bim. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Oberlton, B.; Matulis, S.; Boise, L.H. Induction of Bim-Dependent and-Independent Apoptosis in Multiple Myeloma. Am. Soc. Hematol. 2014, 124, 4716. [Google Scholar]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef]
- Shukla, S.; Saxena, S.; Singh, B.K.; Kakkar, P. BH3-only protein BIM: An emerging target in chemotherapy. Eur. J. Cell Biol. 2017, 96, 728–738. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2009, 27, S71. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Ploner, C.; Kofler, R.; Villunger, A. Noxa: At the tip of the balance between life and death. Oncogene 2009, 27, S84. [Google Scholar] [CrossRef]
- Puthalakath, H.; Villunger, A.; O’Reilly, L.A.; Beaumont, J.G.; Coultas, L.; Cheney, R.E.; Huang, D.C.; Strasser, A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001, 293, 1829–1832. [Google Scholar] [CrossRef]
- Pinon, J.D.; Labi, V.; Egle, A.; Villunger, A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene 2008, 27 (Suppl. 1), S41–S52. [Google Scholar] [CrossRef]
- Atlas, T.H.P. BMF. Available online: https://www.proteinatlas.org/ENSG00000104081-BMF/pathology (accessed on 3 December 2018).
- Shao, Y.; Aplin, A.E. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ. 2012, 19, 2029–2039. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wu, M. p53 and Bad: Remote strangers become close friends. Cell Res. 2007, 17, 283–285. [Google Scholar] [CrossRef]
- Howells, C.C.; Baumann, W.T.; Samuels, D.C.; Finkielstein, C.V. The Bcl-2-associated death promoter (BAD) lowers the threshold at which the Bcl-2-interacting domain death agonist (BID) triggers mitochondria disintegration. J. Theor. Biol. 2011, 271, 114–123. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Bernabeu, A.; Guillen, J.; Perez-Berna, A.J.; Moreno, M.R.; Villalain, J. Structure of the C-terminal domain of the pro-apoptotic protein Hrk and its interaction with model membranes. Biochim. Biophys. Acta 2007, 1768, 1659–1670. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Ortis, F.; Cunha, D.; Gosset, G.; Li, M.; Cardozo, A.K.; Eizirik, D.L. Signaling by IL-1β+ IFN-γ and ER stress converge on DP5/Hrk activation: A novel mechanism for pancreatic β-cell apoptosis. Cell Death Differ. 2009, 16, 1539. [Google Scholar] [CrossRef]
- Chinnadurai, G.; Vijayalingam, S.; Rashmi, R. BIK, the founding member of the BH3-only family proteins: Mechanisms of cell death and role in cancer and pathogenic processes. Oncogene 2008, 27 (Suppl. 1), S20–S29. [Google Scholar] [CrossRef]
- Germain, M.; Mathai, J.P.; Shore, G.C. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 2002, 277, 18053–18060. [Google Scholar] [CrossRef]
- Reed, J.C. Bcl-2 on the brink of breakthroughs in cancer treatment. Cell Death Differ. 2018, 25, 3–6. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Ni Chonghaile, T.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27 (Suppl. 1), S149–S157. [Google Scholar] [CrossRef]
- Redondo, M.; Esteban, F.; Gonzalez-Moles, M.A.; Delgado-Rodriguez, M.; Nevado, M.; Torres-Munoz, J.E.; Tellez, T.; Villar, E.; Morell, M.; Petito, C.K. Expression of the antiapoptotic proteins clusterin and bcl-2 in laryngeal squamous cell carcinomas. Tumour Biol. 2006, 27, 195–200. [Google Scholar] [CrossRef]
- Lickliter, J.D.; Cox, J.; McCarron, J.; Martinez, N.R.; Schmidt, C.W.; Lin, H.; Nieda, M.; Nicol, A.J. Small-molecule Bcl-2 inhibitors sensitise tumour cells to immune-mediated destruction. Br. J. Cancer 2007, 96, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.K.; Sasaki, C.Y.; Hardwick, J.M.; Longo, D.L. Bcl-2–mediated drug resistance: Inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J. Exp. Med. 1999, 190, 253–266. [Google Scholar] [CrossRef]
- Sartorius, U.A.; Krammer, P.H. Upregulation of bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int. J. Cancer 2002, 97, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, M.; Strasser, A. Caspases, Bcl-2 Family Proteins and Other Components of the Death Machinery: Their Role in the Regulation of the Immune Response; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Zheng, Q.; Wang, B.; Gao, J.; Xin, N.; Wang, W.; Song, X.; Shao, Y.; Zhao, C. CD155 knockdown promotes apoptosis via AKT/Bcl-2/Bax in colon cancer cells. J. Cell. Mol. Med. 2018, 22, 131–140. [Google Scholar] [CrossRef]
- Yu, S.; Gong, L.S.; Li, N.F.; Pan, Y.F.; Zhang, L. Galangin (GG) combined with cisplatin (DDP) to suppress human lung cancer by inhibition of STAT3-regulated NF-kappaB and Bcl-2/Bax signaling pathways. Biomed. Pharmacother. 2018, 97, 213–224. [Google Scholar] [CrossRef]
- Timucin, A.C.; Basaga, H.; Kutuk, O. Selective targeting of antiapoptotic BCL-2 proteins in cancer. Med. Res. Rev. 2018. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Garcia-Aranda, M.; Redondo, M. Protein Kinase Targets in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2543. [Google Scholar] [CrossRef]
- Lochmann, T.L.; Bouck, Y.M.; Faber, A.C. BCL-2 inhibition is a promising therapeutic strategy for small cell lung cancer. Oncoscience 2018, 5, 218–219. [Google Scholar]
- Bodur, C.; Basaga, H. Bcl-2 inhibitors: Emerging drugs in cancer therapy. Curr. Med. Chem. 2012, 19, 1804–1820. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Min, Q.; Palida, A.; Zhang, Y.; Tang, R.; Chen, L.; Li, H. 8-Chrysoeriol, as a potential BCL-2 inhibitor triggers apoptosis of SW1990 pancreatic cancer cells. Bioorg. Chem. 2018, 77, 478–484. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves New Drug for Chronic Lymphocytic Leukemia in Patients with a Specific Chromosomal Abnormality. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm495253.htm (accessed on 11 April 2016).
- Begley, J.; Vo, D.D.; Morris, L.F.; Bruhn, K.W.; Prins, R.M.; Mok, S.; Koya, R.C.; Garban, H.J.; Comin-Anduix, B.; Craft, N.; et al. Immunosensitization with a Bcl-2 small molecule inhibitor. Cancer Immunol. Immunother. 2009, 58, 699–708. [Google Scholar] [CrossRef]
- Farsaci, B.; Sabzevari, H.; Higgins, J.P.; Di Bari, M.G.; Takai, S.; Schlom, J.; Hodge, J.W. Effect of a small molecule BCL-2 inhibitor on immune function and use with a recombinant vaccine. Int. J. Cancer 2010, 127, 1603–1613. [Google Scholar] [CrossRef]
- Kim, P.S.; Schlom, J. Potential utility of the pan-Bcl-2 inhibitor GX15–070 (obatoclax) in cancer immunotherapy. OncoImmunology 2014, 3, e29351. [Google Scholar] [CrossRef]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res. 2018, 137, 37–75. [Google Scholar]
- Hu, Y.; Yague, E.; Zhao, J.; Wang, L.; Bai, J.; Yang, Q.; Pan, T.; Zhao, H.; Liu, J.; Zhang, J. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. 2018, 423, 47–59. [Google Scholar] [CrossRef]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Applegate, A.J.; et al. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594–84607. [Google Scholar] [CrossRef]
- Soderquist, R.S.; Crawford, L.; Liu, E.; Lu, M.; Agarwal, A.; Anderson, G.R.; Lin, K.H.; Winter, P.S.; Cakir, M.; Wood, K.C. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat. Commun. 2018, 9, 3513. [Google Scholar] [CrossRef]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Ruvolo, V.R.; Wei, J.; Konopleva, M.; Reed, J.C.; Pellecchia, M.; Andreeff, M.; Ruvolo, P.P. Inhibition of Mcl-1 with the pan–Bcl-2 family inhibitor (–) BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood 2015. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Lartigue, L.; Perkins, G. Targeting Mcl-1 and other Bcl-2 family member proteins in cancer therapy. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef]
- Noll, T.; Schultze-Seemann, S.; Kuckuck, I.; Michalska, M.; Wolf, P. Synergistic cytotoxicity of a prostate cancer-specific immunotoxin in combination with the BH3 mimetic ABT-737. Cancer Immunol. Immunother. 2018, 67, 413–422. [Google Scholar] [CrossRef]
- Li, X.; He, J.; Li, B.; Gao, M.; Zeng, Y.; Lian, J.; Shi, C.; Huang, Y.; He, F. The PPARgamma agonist rosiglitazone sensitizes the BH3 mimetic (-)-gossypol to induce apoptosis in cancer cells with high level of Bcl-2. Mol. Carcinog. 2018, 57, 1213–1222. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Chang, H.; Zou, Z.; Xing, D. Downregulation of MCL-1 and upregulation of PUMA using mTOR inhibitors enhance antitumor efficacy of BH3 mimetics in triple-negative breast cancer. Cell Death Dis. 2018, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Talhi, O.; Jang, D.; Cerella, C.; Gaigneaux, A.; Kim, K.W.; Lee, J.W.; Dicato, M.; Bachari, K.; Han, B.W.; et al. Cytostatic hydroxycoumarin OT52 induces ER/Golgi stress and STAT3 inhibition triggering non-canonical cell death and synergy with BH3 mimetics in lung cancer. Cancer Lett. 2018, 416, 94–108. [Google Scholar] [CrossRef]
- Inao, T.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Kotani, H.; Harada, M. Bcl-2 inhibition sensitizes triple-negative human breast cancer cells to doxorubicin. Oncotarget 2018, 9, 25545–25556. [Google Scholar] [CrossRef]
- Vervloessem, T.; Kerkhofs, M.; La Rovere, R.M.; Sneyers, F.; Parys, J.B.; Bultynck, G. Bcl-2 inhibitors as anti-cancer therapeutics: The impact of and on calcium signaling. Cell Calcium 2018, 70, 102–116. [Google Scholar] [CrossRef]
- Nougarede, A.; Rimokh, R.; Gillet, G. BH4-mimetics and -antagonists: An emerging class of Bcl-2 protein modulators for cancer therapy. Oncotarget 2018, 9, 35291–35292. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, B.; Wang, Y.; Li, Z.; Wu, J.; Yang, Y.; Wei, Y.; Peng, X.; Chen, H.; Chen, R.; et al. miR-216a-5p inhibits malignant progression in small cell lung cancer: Involvement of the Bcl-2 family proteins. Cancer Manag. Res. 2018, 10, 4735–4745. [Google Scholar] [CrossRef]
- Wang, Y.B.; Zhao, X.H.; Li, G.; Zheng, J.H.; Qiu, W. MicroRNA-184 inhibits proliferation and promotes apoptosis of human colon cancer SW480 and HCT116 cells by downregulating C-MYC and BCL-2. J. Cell. Biochem. 2018, 119, 1702–1715. [Google Scholar] [CrossRef]
- Wang, X.; Xie, Y.; Wang, J. Overexpression of MicroRNA-34a-5p Inhibits Proliferation and Promotes Apoptosis of Human Cervical Cancer Cells by Downregulation of Bcl-2. Oncol. Res. 2018, 26, 977–985. [Google Scholar] [CrossRef]
- Villanova, L.; Careccia, S.; De Maria, R.; Fiori, M.E. Micro-Economics of Apoptosis in Cancer: NcRNAs Modulation of BCL-2 Family Members. Int. J. Mol. Sci. 2018, 19, 958. [Google Scholar] [CrossRef]
- Meng, X.; Fu, R. miR-206 regulates 5-FU resistance by targeting Bcl-2 in colon cancer cells. Onco Targets Ther. 2018, 11, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.N.; Qie, P.; Yang, G.; Song, Y.B. miR-181b inhibits chemoresistance in cisplatin-resistant H446 small cell lung cancer cells by targeting Bcl-2. Arch. Med. Sci. 2018, 14, 745–751. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Site of Action | Examples | |
|---|---|---|---|
| Antimetabolites | Interfere with DNA/RNA synthesis by inhibiting purine ring synthesis, ribonucleotide reductase or DNA monomers synthesis normally causing cell death during the S phase of cell growth. | Purines and pyrimidines synthesis | Thioguanine Mercaptopurine Methotrexenate |
| Ribonucleotides | Hydroxycarbamide | ||
| DNA monomers | Methotrexate 5-Fluorouracil | ||
| DNA synthesis | Cytarabine Bleomycin | ||
| Intercalating agents | Interfere with DNA/RNA synthesis, preventing cell duplication | Topoisomerase inhibitors (Etoposide Topotecan, Irinotecan) Antibiotics (anthracyclines, Chromomycin, Doxorubicin) | |
| Cross-linking agents | Cross-link two DNA bases together preventing DNA from being separated during DNA synthesis or transcription. Nucleotide mispairing, leading to mutations. | Alkylating agents Nitrosoureas Platinum-based coordination complexes | |
| Enzyme | Protein synthesis inhibition, leading to cell death by apoptosis | Protein synthesis | L-Asparaginase |
| Microtubule damaging agents | Anti-mitotic agents that inhibit cell proliferation by disrupting the normal function of the mitotic spindle. | Microtubules | Taxanes Vinca alkaloids Taxol Colchicine |
| Enzyme inhibitors | Interfere with normal cell metabolism, leading to cell death | Enzyme activity | Methotrexate Kinase inhibitors |
| Angiogenesis inhibitors | Inhibit endothelial cells’ proliferation and tumor growth | Angiogenesis | Endostatin |
| Classification | Overview | |
|---|---|---|
| Tumor-targeting immunotherapy | Naked monoclonal antibodies | Bind and alter the signaling pathways required by malignant cells’ survival or progression. |
| Activate lethal receptors expressed on the surface of cancer cells. | ||
| Opsonizing antibodies that bind to specific tumor-associated antigens. | ||
| Conjugated monoclonal antibodies | Tumor antigen-associated antibodies coupled with toxins or radionuclides. | |
| Bi-specific T-cell engagers that enhance immune response. | ||
| Oncolytic viruses | Non-pathogenic viral strains that infect and directly or indirectly lead to cancer cells’ death. | |
| Anticancer vaccines | Peptide- and DNA-based vaccines to enhance the ability of resident antigen-presenting cells to present tumor-associated antigens, which activates the host immune system against tumor cells. | |
| Isolation, ex vivo amplification/differentiation/activation and administration of dendritic cells which engages the host immune system against tumor cells. | ||
| Administration of immunomodulatory cytokines, generally as adjuvants for other anticancer treatments. | ||
| Administration of immunomodulatory antibodies such as checkpoint blockers or those that interact with soluble or cellular components of the immune system and activate the immune response. | ||
| Administration of inhibitors of immunosuppressive metabolism which alters cancer cells’ microenvironment with antineoplastic effects. | ||
| Pattern recognition receptors-agonists which activates signal transduction cascades with pro-inflammatory effects that include the activation and secretion of immunostimulatory cytokines, dendritic cells maturation and macrophages/natural-killer cells activation. | ||
| Immunogenic cell death inducers, such as some conventional chemotherapeutics, that stimulate the release of damage-associated molecular patterns by cancer cells, which enhances the activation and maturation of antigen-presenting cells. | ||
| Adoptive cell immunotherapy | Collection, ex vivo selection/modification/expansion/activation and administration of circulating or tumor-infiltrating lymphocytes. | |
| Administration of genetically modified T-cells with enhanced proliferative potential and persistence, unique antigen specificity or improved secretory profile, tumor-infiltrating capacity or cytotoxicity. | ||
| Caspase group | Members | Overview |
|---|---|---|
| Initiator caspases | Caspase-2,-8,-9,-10 | These caspases are at the top of the caspase signaling cascade, being responsible for executioner caspases proteolytic activation during apoptosis [11]. Initiator caspases are characterized by the presence of an extended N-terminal pro-domain essential for their function [11]. |
| Effector or executioner caspases | Caspase-3,-6,-7 | Effector caspases [9,12,13,14] induce biochemical and morphological changes in the cell such as chromatin condensation, DNA and nuclear fragmentation, cytoskeletal and nuclear protein degradation, crosslinking of proteins, formation of apoptotic bodies and expression of ligands for phagocytic cell receptors [9]. |
| Inflammatory caspases | Caspase-1,-4,-5,-11 | Key regulators of inflammation and cell death by inducing pyroptosis and the extracellular release of pro-inflammatory cytokines and danger signals [15]. |
| Other caspases | Caspase-12 caspase-13 and caspase-14 | These caspases are not well characterized and are still under study. Caspase-12 has a role during endoplasmic-specific apoptosis [9]. |
| Pathway | Activation | Description |
|---|---|---|
| Mitochondrial pathway | As a response to different stress signals such as DNA damage, chemotherapeutic agents or ultraviolet (UV) light. | Triggers mitochondrial membrane destabilization and permeabilization, with the subsequent release of pro-apoptotic intermembrane itochondrial factors, such as the enzyme cytochrome C oxidase [12,17], which leads to apoptosome formation, caspase-9 activation [18] and cell death. |
| Lysosomal pathway | As a response to oxidative stress, death-receptors activation, viral proteins and other death stimuli [25]. | Lysosomal membrane permeabilization triggers the release of cathepsins into the cytoplasm, outer mitochondria membrane destabilization and subsequent cytochrome-c release [17,26]. |
| PIDD-osome pathway | As a response to DNA damage [27]. | p53 tumor suppressor gene product (p53)-induced proteins with a death domain (PIDD) can activate caspase-2 and the caspase proteolytic cascade leading to the final execution pathway [27]. |
| Endoplasmic reticulum pathway | Under study. | Sensitizes mitochondria to both extrinsic and intrinsic death signals as well as by initiating cell death signals [28]. |
| BCL-2 Subfamily | Members | BH Domains | Overview | |
|---|---|---|---|---|
| Anti-apoptotic | Bcl-2 | BH1 BH2 BH3 BH4 TM | Bcl-2 is constitutively bound to mitochondrial and/or endoplasmic reticulum membranes and represents the main pro-survival member of the Bcl-2 family [41]. Bcl-2 can sequester activator and sensitizer BH3-only proteins and is also able to bind to the inositol trisphosphate receptors (InsP3R), membrane glycoprotein complexes acting as membrane calcium channels on the endoplasmic reticulum, inhibiting the initiation phase of calcium-mediated apoptosis [42]. Bcl-2 over-expression has been widely described in different types of malignancies and is related to tumor formation, progression, therapy resistance and poorer overall survival [39,43,44]. Although negative Bcl-2 expression has been proposed as a marker of good chemotherapy response in breast cancer patients [45], recent studies have shown Bcl-2 over-expression as a good prognosis factor in patients with different types of cancer including breast [46], colorectal, renal and advanced non-small cell lung cancer [47,48,49]. | |
| B-cell lymphoma-extra large (Bcl-XL) | BH1 BH2 BH3 BH4 TM | Bcl-XL is a transmembrane protein localized in the outer mitochondrial and nucleus membranes, where it may bind to nuclear proteins and modulate transcription factors activity [36]. Bcl-XL can also sequester cytoplasmic p53, inhibiting cell death [50]. Bcl-XL, is usually over-expressed in different types of cancer and has been related to cancer cell growth, migration, invasion, maintenance of cancer stem cell phenotype, angiogenesis, enhanced aggressiveness [36] and apoptosis resistance [36,39]. | ||
| Bcl-2-like protein 2 (Bcl2l2, Bcl-W) | BH1 BH2 BH3 BH4 TM | The active Bcl-W isoform is loosely attached to mitochondria and can be neutralized by BH3 (only) proteins by enhancing the insertion of its C-terminal domain into the membrane [51]. Bcl-W over-expression has been related to different malignancies, including lymphoma, colorectal cancer and gastric cancer [52,53,54,55] and to a worse prognosis [54]. Bcl-W expression is regulated by MYC transcription factor through a specific microRNA [54] and its amount within the cell is modulated by the Akt serine-threonine kinase [56]. | ||
| Induced myeloid leukemia cell differentiation protein (Mcl-1) | BH1 BH2 BH3 BH4 TM | Mcl-1 binds to pro-apoptotic Bim [17], Bak and Bax [57] proteins to prevent apoptosis. Mcl-1-Bim complexes can be cleaved by granzyme B, allowing outer mitochondrial membrane permeabilization and apoptosis [17]. Mcl-1 is highly over-expressed in cancer cells [18] and has been related to chemotherapy-resistance [39]. | ||
| Bcl-2 related protein A1 (Bcl2A1), Bfl-1 | BH1 BH2 BH3 BH4 | In response to apoptotic stimuli, Bcl2A1 can translocate from the mitochondria or the cytoplasm to the nucleus [58] where its role remains unclear. Similarly to Mcl1, Bcl2A1 pro-survival ability is related to its association to pro-apoptotic BH3-only Bim, Bid and Puma proteins as well as to Bik, Hrk and Noxa [18]. Although Bcl2A1 is usually over-expressed in cancer cells [18] and contributes to the acquisition of tumor cell resistance against chemotherapy-induced apoptosis [58], the role of Bcl2A1 in both healthy and cancer cells is still under study [58]. Bcl2A1 is regulated at post-translational level by the proteasome and by transcription factors such as NFĸB [58] or retinoic acid [18]. | ||
| Bcl-B Bcl2l10 | BH1 BH2 BH3 BH4 TM | Bcl-B binds to Bcl-2, bcl-XL and Bax, but not to Bak, and is able to suppress Bax-induced apoptosis in vitro [59]. Bcl-B is over-expressed in multiple-myeloma patients [60]. | ||
| Pro-apoptotic | Effectors | Bcl-2-associated X protein (Bax) | BH1 BH2 BH3 TM | Along with Bak, Bax is one of the main apoptosis effectors. Bax exists as a free inactive cytosolic protein that responds to various stimuli exposing the BH3 domain to allow oligomerization [23] and then migrating and inserting into the mitochondria membrane, inducing the release of cytochrome-c [30]. Bax activity is mainly regulated by the cytosolic accumulation of the tumor suppressor protein p53 [61] as well as by other Bcl-2 family members [23]. |
| Bcl-2 homologous antagonist killer (Bak) | BH1 BH2 BH3 TM | Bak, is one of the main apoptosis effectors. After activation by stress signals, this integral mitochondrial membrane protein is activated by exposing the BH3 domain to allow oligomerization and outer mitochondrial membrane destabilization [23]. Bak can directly be activated by the tumor suppressor p53 by blocking the Mcl1 anti-apoptotic effect [62] and can also be regulated by other Bcl-2 family members [23]. | ||
| Bcl-2 related ovarian killer (Bok) | BH1 BH2 BH3 TM | Contrary to Bax or Bak, Bok is constitutively active and unresponsive to the inhibitory effects of Bcl-2 anti-apoptotic members [63], being able to trigger mitochondrial membrane permeabilization and apoptosis independently of Bax and Bak presence [63]. Bok activity, which is controlled by ubiquitylation and proteasomal degradation [63], is an essential mediator of p53-dependent apoptosis [64]. | ||
| Activators | BH3-interacting domain death agonist (Bid) | BH3 | Bid responds to tumor suppressor p53, contributing to cell death as response to cell damage after chemotherapy [65,66]. On the other hand, Bid can also be cleaved and activated by granzyme B [17] as well as by Caspase-8 after death receptor signaling (Fas-ligation-mediated apoptosis). For these reasons, Bid has a key role as a connecting element between the intrinsic and the extrinsic apoptosis pathways [67]. After activation, Bid exposes the BH3 domain which allows its dimerization with apoptosis-effectors Bax, Bak and anti-apoptotic Bcl-2-like proteins [23], resulting in Bax and Bak activation and Bcl-2-like proteins inhibition and subsequent cell death. Once activated, Bid can also migrate from cytosol to mitochondria where it can directly promote the release of cytochrome c and other apoptogenic factors [17,68], amplifying caspase activation. Low Bid expression is related to resistance to chemotherapy [69] and TRAIL [70]. | |
| Bcl-2-like protein 11 (Bim) | BH3 TM | Bim can appear associated to microtubules [67] or sequestered forming complexes with all pro-survival proteins [23]. These complexes can be disrupted by tumor suppressor p53 [71] as a response to cellular stress [23] and also by Granzyme B [17], allowing Bim activation and translocation to mitochondrial outer membrane to indirectly cause cell death by pro-apoptotic Bak/Bax activation [67,72,73]. Bim expression is regulated at different levels, and its abundance is controlled via the proteasome by protein kinases downstream growth factor receptor activation [67]. Bim has been reported to play a central role in regulation of tumorigenesis [74]. Indeed, Bim over-expression inhibits tumor growth and drug resistance [74], while Bim loss is associated with lymphadenopathy, autoimmunity [67] and tumor promotion [74]. | ||
| p53 upregulated modulator of apoptosis (Puma) | BH3 | Similarly to Bid and Bim, Puma can directly bind and antagonize all pro-survival proteins [23,75] by directly or indirectly promoting cell death [75,76]. Puma, whose expression can be induced by nuclear p53 [50,76] after cellular stress or DNA damage [23,50,77], is able to displace cytoplasmic p53 from anti-apoptotic Bcl-xL, allowing p53 to induce cell death [50]. Puma expression can also be activated by transcription factors induced as a response to stimuli such as genotoxic stress, deregulated oncogene expression or toxins, being able to induce cell death in a p53-independent manner [75]. Puma, which is required by Bad and Noxa to induce cell death [73], can also directly activate pro-apoptotic Bax and Bak to promote mitochondrial cytochrome c release [73]. Aberrant Puma expression has been related to increased cancer risk development and therapeutic resistance [67,75]. | ||
| Bcl2 like 11, Bcl2 modifying factor (Bmf) | BH3 | Similar to Bim, Bmf is bound to cytoskeletal structures [67,78]. After cellular damage or anoikis, Bmf is unleashed, being able to sequester pro-survival Bcl-2 proteins and promote cell death [78]. Bmf, which is widely expressed in lymphocytes and most hematopoietic tissues [79], is also expressed in different malignancies [80] including lung and breast [79]. Aberrant Bmf expression has been related to acquired resistance to chemotherapy [81]. | ||
| Sensitizers | Bcl-2-associated death promoter (Bad) | BH3 | Bad promotes apoptosis by interacting with and inhibiting the anti-apoptotic function of Bcl-2 and Bcl-XL [82] and also, by sensitizing the cell to Bid-induced mitochondria disintegration [83]. Bad activity is determined by its hosphorylation status [82], which is modulated by protein kinases, including Akt [82] downstream growth factor receptor activation [67], allowing Bad dimerization and its sequestration away from the mitochondria [30]. As a response to death stimuli, Bad is dephosphorylated and translocated to the mitochondria to induce cell death [82]. In this regard, recent studies have shown that tumor suppressor p53 can strongly bind to dephosphorylated Bad, which determines Bad pro-apoptotic role [82]. The association between Bad and p53 also regulates Bad expression, by preventing p53 entrance into the nucleus to bind Bad promoter [82]. | |
| Noxa (Damage in Latin) | BH3 | Noxa is associated to the mitochondrial membrane [84] and can be activated, by exposing its BH3 domain and disrupting mitochondrial membrane [84] in response to cellular stress [77], such as DNA-damage [67], in a p53-dependent or -independent manner [77]. Noxa activation sensitizes the cell toward the action of other BH3-only proteins [77] and enhances the activation of Bax and/or Bak [77]. Noxa can also bind to anti-apoptotic members of Bcl-2 family [84], such as Mcl1, for proteasomal degradation [77], being also able to neutralize the pro-apoptotic effect of Bcl-XL [41]. | ||
| Harakiri (Hrk) DP5 | BH3 TM | Hrk is associated to the mitochondria and is able to promote apoptosis via mitochondrial outer membrane permeabilization [85] as a response to different signals such as endoplasmic reticulum stress or cytokines [86]. Hrk is regulated by the interaction with the apoptotic inhibitors Bcl-2 and Bcl-X(L) via its BH3 domain [38,85] and its loss contributes to neoplasia and autoimmunity [85]. | ||
| Bcl-2-interacting killer (Bik) | BH3 TM | Bik localizes in the endoplasmic reticulum outer membrane [87]. As a response to stress signals and Bax activation, Bik promotes apoptosis mobilizing calcium to the mitochondria, remodeling the mitochondrial cristae [87] and provoking cytochrome c release [88]. BIK has been proposed as a new target for anti-cancer drugs that inhibit proteasomal functions as well as for the treatment of difficult cancers [87]. | ||
| Type | Small Molecule Inhibitor | Disease | |
|---|---|---|---|
| Phase I | Mcl-1 protein inhibitor | AMG-176 AZD-5991 | Multiple myeloma |
| Hematological cancer | |||
| S-64315 (MIK-665) | Diffuse large B-cell lymphoma, multiple myeloma | ||
| S-64315 (MIK-665) | Myelodysplastic syndrome, Acute myeloid leukemia (AML) | ||
| Bcl-2 protein inhibitor | Venetoclax | Non-Hodgkin lymphoma, myelodysplastic syndrome | |
| BCL-201 (S-55746) | Mantle cell lymphoma, follicular lymphoma | ||
| BCL-201 (S-55746) | Myelodysplastic syndrome, CLL, AML, NHL | ||
| Bcl-2, Bcl-xL inhibitor | APG-1252 | Tumor, small cell lung cancer | |
| AT-101 | Multiple myeloma | ||
| Navitoclax | Acute lymphoblastic leukemia | ||
| Phase II | Bcl-2 protein inhibitor | Venetoclax | Diffuse large B cell lymphoma, B cell lymphoma, myelodysplastic syndrome (suspended), follicular lymphoma |
| Venetoclax | DLBCL, B cell lymphoma, myelodysplastic syndrome (suspended), Waldenström macroglobulinemia, hematological neoplasm, follicular lymphoma | ||
| Bcl-2, Bcl-xL inhibitor | Navitoclax | Myelofibrosis | |
| Phase III | Bcl-2 protein inhibitor | Venetoclax | Multiple myeloma, acute myeloid leukemia |
| Venetoclax | Multiple myeloma, AML, mantle cell lymphoma | ||
| Venetoclax | Chronic lymphocytic leukemia [31] |
| Inhibitor | Cancer Models |
|---|---|
| HA14-1 and derivatives | Lymphoma, leukemia, myeloma, glioma, ovarian, prostate |
| BH3Is | Leukemia, cervical, glioma |
| 2-Methoxy-antimycin A3 | Lung, mesothelioma, esophageal |
| (-)-Gossypol | Lymphoma1, leukemia2, myeloma3, prostate, colon, head and neck squamous cell carcinoma |
| TW-37 | Lymphoma, leukemia, prostate, lung, pancreatic, liver, nasopharyngeal |
| Apogossypolone and derivatives | Lymphoma, leukemia, prostate, lung, pancreatic, liver, nasopharyngeal |
| BI-97CI | Lymphoma, prostate, lung |
| Chelerythrine | Leukemia, liver, cardiac, neuroblastoma |
| YC137 | Breast |
| Obatoclax | Lymphoma, leukemia, myeloma, lung, mammary carcinoma, colon, cervical, prostate |
| ABT-737/263 | Lymphoma, leukemia, myeloma, prostate, lung, pancreatic, ovarian, colorectal, gastrointestinal |
| S1 | Breast, liver, cervical |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950. https://doi.org/10.3390/ijms19123950
García-Aranda M, Pérez-Ruiz E, Redondo M. Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. International Journal of Molecular Sciences. 2018; 19(12):3950. https://doi.org/10.3390/ijms19123950
Chicago/Turabian StyleGarcía-Aranda, Marilina, Elisabet Pérez-Ruiz, and Maximino Redondo. 2018. "Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy" International Journal of Molecular Sciences 19, no. 12: 3950. https://doi.org/10.3390/ijms19123950
APA StyleGarcía-Aranda, M., Pérez-Ruiz, E., & Redondo, M. (2018). Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. International Journal of Molecular Sciences, 19(12), 3950. https://doi.org/10.3390/ijms19123950