The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies


 , and
, and
Abstract
:
1. Introduction
2. Results
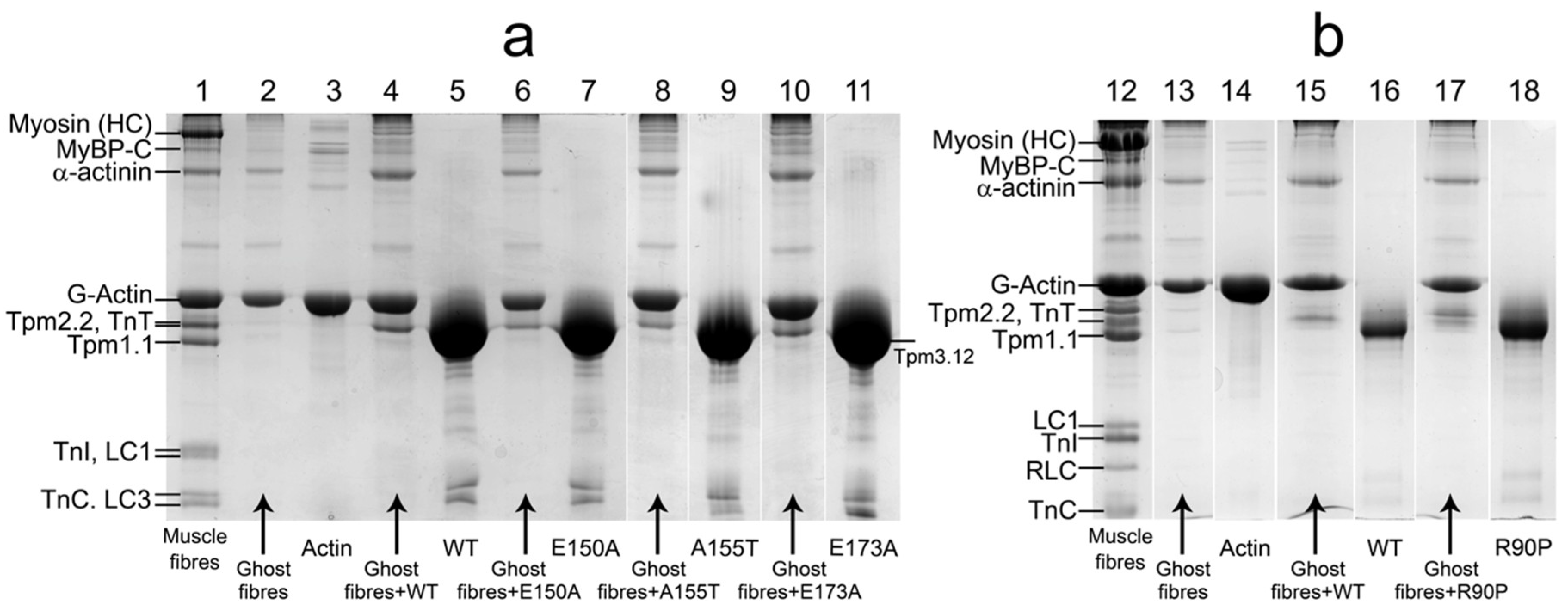
2.1. Ghost Muscle Fibers Reconstituted with Labeled Proteins as a Model for Study of the Conformational Changes in Proteins During Muscle Contraction
2.2. The Effect of Ca2+ and Nucleotides on the Conformational State of Wild-Type γ-Tropomyosin, Actin, and on the Binding of Myosin Heads to F-Actin during the ATPase Cycle
2.3. The Effect of the E173A, R90P, E150A, and A155T Mutations in γ-Tropomyosin on its Position and Rigidity in Unregulated Actin Filaments
2.4. The E173A, R90P, E150A, and A155T Mutations in Tpm3.12 Increase the Ca2+-Sensitivity of the Actin-Activated ATPase Activity of Myosin Sufragment-1
2.5. The Effect of the E173A, R90P, E150A, and A155T Mutations in γ-Tropomyosin on Conformational State of Actin and Tpm, and on the Binding of Myosin Heads to F-Actin at Low Ca2+ During the ATPase Cycle
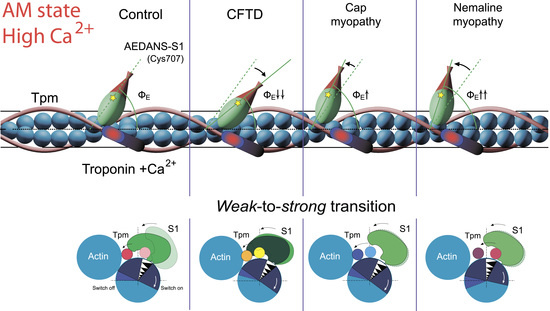
2.6. The Effect of the E173A, R90P, E150A, and A155T Mutations in γ-Tpm on the Conformational State of Actin and Tpm, and on the Binding of Myosin Heads to F-Actin at High Ca2+ During the ATPase Cycle
3. Discussion
4. Materials and Methods
4.1. Chemical and Solutions
4.2. The Rabbit Muscle Proteins Preparation
4.3. The Recombinant γγ-Tpm Obtaining
4.4. ATPase Measurements
4.5. Ghost Fibers Preparation and Reconstruction of Regulatory and Contractile System
4.6. Electrophoretic Separation of Muscle Fiber Proteins
4.7. Fluorescence Polarization Measurement
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CFTD | Congenital Fiber-Type Disproportion |
| NM | Nemaline Myopathy |
| DA | Distal Arthrogryposis |
| Cap | Cap myopathy |
| Tpm | Tropomyosin |
| TN | Troponin |
| S1 | Myosin Subfragment-1 |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| 5-IAF | 5-Iodoacetoamidofluorescein |
| 1,5-IAEDANS | 5-({2-[(iodoacetyl)amino]ethyl}amino)naphthalene-1-sulfonic acid |
References
- Rynkiewicz, M.J.; Prum, T.; Hollenberg, S.; Kiani, F.A.; Fagnant, P.M.; Marston, S.B.; Trybus, K.M.; Fischer, S.; Moore, J.R.; Lehman, W. Tropomyosin Must Interact Weakly with Actin to Effectively Regulate Thin Filament Function. Biophys. J. 2017, 113, 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Lehman, W. Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments. J. Mol. Biol. 2001, 311, 1027–1036. [Google Scholar] [CrossRef]
- Lehman, W.; Li, X.; Kiani, F.A.; Moore, J.R.; Campbell, S.G.; Fischer, S.; Rynkiewicz, M.J. Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophys. J. 2018, 115, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Redwood, C.S. Modulation of the effects of tropomyosin on actin and myosin conformational changes by troponin and Ca2+. Biochim. Biophys. Acta Proteins Proteom. 2009, 1794, 985–994. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Rysev, N.A.; Karpicheva, O.E.; Sirenko, V.V.; Avrova, S.V.; Piers, A.; Redwood, C.S. Molecular mechanisms of dysfunction of muscle fibres associated with Glu139 deletion in TPM2 gene. Sci. Rep. 2017, 7, 16797. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Simonyan, A.O.; Chernev, A.A.; Karpicheva, O.E.; Piers, A.; Redwood, C.S. Aberrant movement of β-tropomyosin associated with congenital myopathy causes defective response of myosin heads and actin during the ATPase cycle. Arch. Biochem. Biophys. 2015, 577–578, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Marttila, M.; Lehtokari, V.L.; Marston, S.; Nyman, T.A.; Barnerias, C.; Beggs, A.H.; Bertini, E.; Ceyhan-Birsoy, O.; Cintas, P.; Gerard, M.; et al. Mutation Update and Genotype-Phenotype Correlations of Novel and Previously Described Mutations in TPM2 and TPM3 Causing Congenital Myopathies. Hum. Mutat. 2014, 35, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Sewry, C.A.; Wallgren-Pettersson, C. Myopathology in congenital myopathies. Neuropathol. Appl. Neurobiol. 2017, 43, 5–23. [Google Scholar] [CrossRef]
- Lehtokari, V.-L.; Ceuterick-de Groote, C.; de Jonghe, P.; Marttila, M.; Laing, N.G.; Pelin, K.; Wallgren-Pettersson, C. Cap disease caused by heterozygous deletion of the beta-tropomyosin gene TPM2. Neuromuscul. Disord. 2007, 17, 433–442. [Google Scholar] [CrossRef]
- Clarke, N.F. Congenital fibre type disproportion: A syndrome at the crossroads of the congenital myopathies. Neuromuscul. Disord. 2011, 21, 252–253. [Google Scholar] [CrossRef]
- Malfatti, E.; Schaeffer, U.; Chapon, F.; Yang, Y.; Eymard, B.; Xu, R.; Laporte, J.; Romero, N.B. Combined cap disease and nemaline myopathy in the same patient caused by an autosomal dominant mutation in the TPM3 gene. Neuromuscul. Disord. 2013, 23, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Tajsharghi, H.; Ohlsson, M.; Lindberg, C.; Oldfors, A. Congenital myopathy with nemaline rods and cap structures caused by a mutation in the beta-tropomyosin gene (TPM2). Arch. Neurol. 2007, 64, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Michele, D.E.; Albayya, F.P.; Metzger, J.M. A nemaline myopathy mutation in α-tropomyosin causes defective regulation of striated muscle force production. J. Clin. Investig. 1999, 104, 1575–1581. [Google Scholar] [CrossRef] [Green Version]
- Miro, O.; Masanes, F.; Pedrol, E.; Garcia-Carrasco, M.; Mallolas, J.; Casademont, J.; Grau, J.M. A comparative study of the clinical and histological characteristics between classic nemaline myopathy and that associated with the human immunodeficiency virus. Med. Clin. 1995, 105, 500–503. [Google Scholar]
- Sun, A.-P.; Ohtsuki, Y.; Yano, T.; Matsumoto, M.; Takeuchi, T.; Furihata, M.; Arahori, M.; Sonobe, H. Typical nemaline bodies presenting in a patient with polymyositis. Med. Electron Microsc. 2002, 35, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, V.; Sewry, C.A. Muscle Biopsy: A Practical Approach, 3rd ed.; Sanders Elsevier: Edinburg, UK, 2007. [Google Scholar]
- Munot, P.; Lashley, D.; Jungbluth, H.; Feng, L.; Pitt, M.; Robb, S.A.; Palace, J.; Jayawant, S.; Kennet, R.; Beeson, D.; et al. Congenital fibre type disproportion associated with mutations in the tropomyosin 3 (TPM3) gene mimicking congenital myasthenia. Neuromuscul. Disord. 2010, 20, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.W.; Dechene, E.T.; Roumm, E.; Geggel, A.S.; Moghadaszadeh, B.; Beggs, A.H. Mutations of tropomyosin 3 (TPM3) are common and associated with type 1 myofiber hypotrophy in congenital fiber type disproportion. Hum. Mutat. 2010, 31, 176–183. [Google Scholar] [CrossRef]
- Kiphuth, I.C.; Krause, S.; Huttner, H.B.; Dekomien, G.; Struffert, T.; Schröder, R. Autosomal dominant nemaline myopathy caused by a novel α-tropomyosin 3 mutation. J. Neurol. 2010, 257, 658–660. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Dedova, I.V.; dos Remedios, C.G.; Vikhoreva, N.N.; Vikhorev, P.G.; Avrova, S.V.; Hazlett, T.L.; van der Meer, B.W. Fluorescence depolarization of actin filaments in reconstructed myofibers: The effect of S1 or pPDM-S1 on movements of distinct areas of actin. Biophys. J. 2004, 86, 3020–3029. [Google Scholar] [CrossRef]
- Borovikov, Y.S. Conformational changes of contractile proteins and their role in muscle contraction. Int. Rev. Cytol. 1999, 189, 267–301. [Google Scholar]
- Roopnarine, O.; Thomas, D.D. Orientation of intermediate nucleotide states of indane dione spin-labeled myosin heads in muscle fibers. Biophys. J. 1996, 70, 2795–2806. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.B.; Lataro, R.C.; Ferro, J.A.; de Reinach, F. Functional alpha-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 1994, 269, 10461–10466. [Google Scholar] [PubMed]
- Borejdo, J.; Assulin, O.; Ando, T.; Putnam, S. Cross-bridge orientation in skeletal muscle measured by linear dichroism of an extrinsic chromophore. J. Mol. Biol. 1982, 158, 391–414. [Google Scholar] [CrossRef]
- Lorenz, M.; Popp, D.; Holmes, K.C. Refinement of the F-actin model against X-ray fiber diffraction data by the use of a directed mutation algorithm. J. Mol. Biol. 1993, 234, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Rysev, N.A.; Nevzorov, I.A.; Avrova, S.V.; Karpicheva, O.E.; Redwood, C.S.; Levitsky, D.I.; Borovikov, Y.S. Gly126Arg substitution causes anomalous behaviour of α-skeletal and β-smooth tropomyosins during the ATPase cycle. Arch. Biochem. Biophys. 2014, 543, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Karpicheva, O.E.; Robinson, P.; Piers, A.; Borovikov, Y.S.; Redwood, C.S. The nemaline myopathy-causing E117K mutation in beta-tropomyosin reduces thin filament activation. Arch. Biochem. Biophys. 2013, 536, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Borejdo, J.; Shepard, A.; Dumka, D.; Akopova, I.; Talent, J.; Malka, A.; Burghardt, T.P. Changes in orientation of actin during contraction of muscle. Biophys. J. 2004, 86, 2308–2317. [Google Scholar] [CrossRef]
- Borejdo, J.; Muthu, P.; Talent, J.; Akopova, I.; Burghardt, T.P. Rotation of actin monomers during isometric contraction of skeletal muscle. J. Biomed. Opt. 2007, 12, 14013. [Google Scholar] [CrossRef]
- Irving, M.; Allen, T.S.C.; Sabido-David, C.; Craik, J.S.; Brandmeier, B.; Kendrick-Jones, J.; Corrie, J.E.; Trentham, D.R.; Goldman, Y.E. Tilting of the light-chain region of myosin during step length changes and active force generation in skeletal muscle. Nature 1995, 375, 688–691. [Google Scholar] [CrossRef]
- Burghardt, T.P.; Cruz-Walker, A.R.; Park, S.; Ajtai, K. Conformation of myosin interdomain interactions during contraction: Deductions from muscle fibers using polarized fluorescence. Biochemistry 2001, 40, 4821–4833. [Google Scholar] [CrossRef]
- Corrie, J.E.; Brandmeier, B.D.; Ferguson, R.E.; Trentham, D.R.; Kendrick-Jones, J.; Hopkins, S.C.; van der Heide, U.A.; Goldman, Y.E.; Sabido-David, C.; Dale, R.E.; et al. Dynamic measurement of myosin light-chain-domain tilt and twist in muscle contraction. Nature 1999, 400, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Titus, M.A.; Ashiba, G.; Szent-Gyorgyi, A.G. SH-1 modification of rabbit myosin interferes with calcium regulation. J. Muscle Res. Cell Motil. 1989, 10, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Bobkov, A.A.; Bobkova, E.A.; Homsher, E.; Reisler, E. Activation of regulated actin by SH1-modified myosin subfragment 1. Biochemistry 1997, 36, 7733–7738. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Nitanai, Y. Thiol reactivity as a sensor of rotation of the converter in myosin. Biochem. Biophys. Res. Commun. 2008, 369, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Isambert, H.; Venier, P.; Maggs, A.C.; Fattoum, A.; Kassab, R.; Pantaloni, D.; Carlier, M.F. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J. Biol. Chem. 1995, 270, 11437–11444. [Google Scholar] [CrossRef] [PubMed]
- Dancker, P.; Low, I.; Hasselbach, W.; Wieland, T. Interaction of actin with phalloidin: Polymerization and stabilization of F-actin. Biochim. Biophys. Acta 1975, 400, 407–414. [Google Scholar] [CrossRef]
- Bukatina, A.E.; Fuchs, F. Effect of phalloidin on the ATPase activity of striated muscle myofibrils. J. Muscle Res. Cell Motil. 1994, 15, 29–36. [Google Scholar] [CrossRef]
- Prochniewicz-Nakayama, E.; Yanagida, T.; Oosawa, F. Studies on conformation of F-actin in muscle fibers in the relaxed state, rigor, and during contraction using fluorescent phalloidin. J. Cell Biol. 1983, 97, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Lamkin, M.; Tao, T.; Lehrer, S.S. Tropomyosin-troponin and tropomyosin-actin interactions: A fluorescence quenching study. Biochemistry 1983, 22, 3053–3058. [Google Scholar] [CrossRef]
- Rynkiewicz, M.J.; Schott, V.; Orzechowski, M.; Lehman, W.; Fischer, S. Electrostatic interaction map reveals a new binding position for tropomyosin on F-actin. J. Muscle Res. Cell Motil. 2015, 36, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Li, X.E.; Holmes, K.C.; Lehman, W.; Jung, H.; Fischer, S. The Shape and Flexibility of Tropomyosin Coiled Coils: Implications for Actin Filament Assembly and Regulation. J. Mol. Biol. 2010, 395, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, M.; Moore, J.R.; Fischer, S.; Lehman, W. Tropomyosin movement on F-actin during muscle activation explained by energy landscapes. Arch. Biochem. Biophys. 2014, 545. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, T.; Taniguchi, M.; Oosawa, F. Conformational changes of F-actin in the thin filaments of muscle induced in vivo and in vitro by calcium ions. J. Mol. Biol. 1974, 90, 509–522. [Google Scholar] [CrossRef]
- Lehman, W.; Orzechowski, M.; Li, X.E.; Fischer, S.; Raunser, S. Gestalt-Binding of tropomyosin on actin during thin filament activation. J. Muscle Res. Cell Motil. 2013, 34, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egelman, E.H.; Francis, N.; DeRosier, D.J. F-actin is a helix with a random variable twist. Nature 1982, 298, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Sugimoto, Y.; Tanaka, H.; Ueno, Y.; Takezawa, Y.; Amemiya, Y. X-ray diffraction evidence for the extensibility of actin and myosin filaments during muscle contraction. Biophys. J. 1994, 67, 2422–2435. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, T.; Oosawa, F. Polarized fluorescence from epsilon-ADP incorporated into F-actin in a myosin-free single fiber: Conformation of F-actin and changes induced in it by heavy meromyosin. J. Mol. Biol. 1978, 126, 507–524. [Google Scholar] [CrossRef]
- Dedova, I.V.; Avrova, S.V.; Vikhoreva, N.N.; Vikhorev, R.G.; Hazlett, T.L.; van der Meer, W.; dos Remedios, C.G.; Borovikov, I.S. Conformational changes of actin induced by strong or weak myosin subfragment-1 binding. Tsitologiia 2004, 46, 719–734. [Google Scholar]
- Borovikov, Y.S. The changes in disposition of various regions of actin monomer and myosin-head during ATP hydrolysis cycle. In Muscle as a Machine. Energy Transduction in the Contractile System; Morales, M., Ed.; National Institutes of Health: Bathesda, MD, USA, 1992; pp. 30–32. [Google Scholar]
- Avrova, S.V.; Rysev, N.A.; Matusovsky, O.S.; Shelud’ko, N.S.; Borovikov, Y.S. Twitchin can regulate the ATPase cycle of actomyosin in a phosphorylation-dependent manner in skinned mammalian skeletal muscle fibres. Arch. Biochem. Biophys. 2012, 521, 1–9. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Resetar, A.M.; Stephens, J.M.; Chalovich, J.M. Troponin-tropomyosin: An allosteric switch or a steric blocker? Biophys. J. 2002, 83, 1039–1049. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Rysev, N.A.; Avrova, S.V.; Karpicheva, O.E.; Borys, D.; Moraczewska, J. Molecular mechanisms of deregulation of the thin filament associated with the R167H and K168E substitutions in tropomyosin Tpm1.1. Arch. Biochem. Biophys. 2017, 614, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.J.; Wang, C.-L.A.; Lehman, W.; Moore, J.R. Modulation of actin mechanics by caldesmon and tropomyosin. Cell Motil. Cytoskeleton. 2008, 65, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Craig, R.; Tobacman, L.; Horowitz, R.; Lehman, W. Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys. J. 1999, 77, 985–992. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Karpicheva, O.E.; Chudakova, G.A.; Robinson, P.; Redwood, C.S. Dilated cardiomyopathy mutations in alpha-tropomyosin inhibit its movement during the ATPase cycle. Biochem. Biophys. Res. Commun. 2009, 381, 403–406. [Google Scholar] [CrossRef]
- Behrmann, E.; Müller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Avrova, S.V.; Karpicheva, O.E.; Robinson, P.; Redwood, C.S. The effect of the dilated cardiomyopathy-causing Glu40Lys TPM1 mutation on actin-myosin interactions during the ATPase cycle. Biochem. Biophys. Res. Commun. 2011, 411, 496–500. [Google Scholar] [CrossRef]
- Karpicheva, O.E.; Sirenko, V.V.; Rysev, N.A.; Simonyan, A.O.; Borys, D.; Moraczewska, J.; Borovikov, Y.S. Deviations in conformational rearrangements of thin filaments and myosin caused by the Ala155Thr substitution in hydrophobic core of tropomyosin. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1790–1799. [Google Scholar] [CrossRef]
- Crick, F.H.C. The packing of α-helices. Simple coiled-coils. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar] [CrossRef]
- McLachlan, A.D.; Stewart, M. Tropomyosin coiled-coil interactions: Evidence for an unstaggered structure. J. Mol. Biol. 1975, 98, 293–304. [Google Scholar] [CrossRef]
- Singh, A.; Hitchcock-DeGregori, S.E. A peek into tropomyosin binding and unfolding on the actin filament. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Marttila, M.; Lemola, E.; Wallefeld, W.; Memo, M.; Donner, K.; Laing, N.G.; Marston, S.; Grönholm, M.; Wallgren-Pettersson, C. Abnormal actin binding of aberrant β-tropomyosins is a molecular cause of muscle weakness in TPM2-related nemaline and cap myopathy. Biochem. J. 2012, 442, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Robaszkiewicz, K.; Dudek, E.; Kasprzak, A.A.; Moraczewska, J. Functional effects of congenital myopathy-related mutations in gamma-tropomyosin gene. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1562–1569. [Google Scholar] [CrossRef]
- Li, X.E.; Lehman, W.; Fischer, S. The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. J. Struct. Biol. 2010, 170, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borovikov, Y.S.; Simonyan, A.O.; Karpicheva, O.E.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Piers, A.; Redwood, C.S. The reason for a high Ca(2+)-sensitivity associated with Arg91Gly substitution in TPM2 gene is the abnormal behavior and high flexibility of tropomyosin during the ATPase cycle. Biochem. Biophys. Res. Commun. 2017, 494, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Karpicheva, O.E.; Simonyan, A.O.; Kuleva, N.V.; Redwood, C.S.; Borovikov, Y.S. Myopathy-causing Q147P TPM2 mutation shifts tropomyosin strands further towards the open position and increases the proportion of strong-binding cross-bridges during the ATPase cycle. Biochim. Biophys. Acta 2016, 1864, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.S.; Brassington, A.-M.E.; Grannatt, K.; Rutherford, A.; Whitby, F.G.; Krakowiak, P.A.; Jorde, L.B.; Carey, J.C.; Bamshad, M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genet. 2003, 72, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Donner, K.; Ollikainen, M.; Ridanpaa, M.; Christen, H.-J.; Goebel, H.H.; de Visser, M.; Pelin, K.; Wallgren-Pettersson, C. Mutations in the beta-tropomyosin (TPM2) gene—A rare cause of nemaline myopathy. Neuromuscul. Disord. 2002, 12, 151–158. [Google Scholar] [CrossRef]
- Brandis, A.; Aronica, E.; Goebel, H.H. TPM2 mutation. Neuromuscul. Disord. 2008, 18, 1005. [Google Scholar] [CrossRef]
- Clarke, N.F.; Domazetovska, A.; Waddell, L.; Kornberg, A.; McLean, C.; North, K.N. Cap disease due to mutation of the beta-tropomyosin gene (TPM2). Neuromuscul. Disord. 2009, 19, 348–351. [Google Scholar] [CrossRef]
- Avrova, S.V.; Karpicheva, O.E.; Rysev, N.A.; Simonyan, A.O.; Sirenko, V.V.; Redwood, C.S.; Borovikov, Y.S. The reason for the low Ca(2+)-sensitivity of thin filaments associated with the Glu41Lys mutation in the TPM2 gene is “freezing” of tropomyosin near the outer domain of actin and inhibition of actin monomer switching off during the ATPase cycle. Biochem. Biophys. Res. Commun. 2018, 502, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Karpicheva, O.E.; Redwood, C.S.; Borovikov, Y.S. The E117K mutation in β-tropomyosin disturbs concerted conformational changes of actomyosin in muscle fibers. Arch. Biochem. Biophys. 2014, 549, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Lu, X.; Suzuki, M.; Ishiwata, S.; Kawai, M. The effect of tropomyosin on force and elementary steps of the cross-bridge cycle in reconstituted bovine myocardium. J. Physiol. 2004, 556, 637–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geeves, M.A.; Holmes, K.C. The molecular mechanism of muscle contraction. Adv. Protein Chem. 2005, 71, 161–193. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Memo, M.; Messer, A.; Papadaki, M.; Nowak, K.; Mcnamara, E.; Ong, R.; El-Mezgueldi, M.; Li, X.; Lehman, W. Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 2013, 22, 4978–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieczorek, D.F.; Jagatheesan, G.; Rajan, S. The role of tropomyosin in heart disease. Adv. Exp. Med. Biol. 2008, 644, 132–142. [Google Scholar] [PubMed]
- Ohlsson, M.; Quijano-Roy, S.; Darin, N.; Brochier, G.; Lacene, E.; Avila-Smirnow, D.; Fardeau, M.; Oldfors, A.; Tajsharghi, H. New morphologic and genetic findings in cap disease associated with beta-tropomyosin (TPM2) mutations. Neurology 2008, 71, 1896–1901. [Google Scholar] [CrossRef] [PubMed]
- Rysev, N.A.; Karpicheva, O.E.; Redwood, C.S.; Borovikov, Y.S. The effect of the Asp175Asn and Glu180Gly TPM1 mutations on actin-myosin interaction during the ATPase cycle. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Robinson, P.; Redwood, C.S. The effect of the dilated cardiomyopathy-causing mutation Glu54Lys of alpha-tropomyosin on actin-myosin interactions during the ATPase cycle. Arch. Biochem. Biophys. 2009, 489, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.; Marston, S.; Willott, R.; Ashley, C.; Mogensen, J.; McKenna, W.; Robinson, P.; Redwood, C.; Watkins, H. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 2005, 280, 28498–28506. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.F.; Kolski, H.; Dye, D.E.; Lim, E.; Smith, R.L.L.; Patel, R.; Fahey, M.C.; Bellance, R.; Romero, N.B.; Johnson, E.S.; et al. Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann. Neurol. 2008, 63, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Site of Stanford University. Available online: https://web.stanford.edu (accessed on 12 September 2018).
- Spudich, J.A.; Watt, S. The regulation of rabbit skeletal muscle contraction. J. Biol. Chem. 1971, 246, 4866. [Google Scholar] [PubMed]
- Margossian, S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzym. 1982, 85, 55–71. [Google Scholar]
- Okamoto, Y.; Sekine, T. A streamlined method of subfragment one preparation from myosin. J. Biochem. 1985, 98, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.D. Preparation of troponin and its subunits. Methods Enzym. 1982, 85, 241–263. [Google Scholar]
- Robinson, P.; Lipscomb, S.; Preston, L.C.; Altin, E.; Watkins, H.; Ashley, C.C.; Redwood, C.S. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007, 21, 896–905. [Google Scholar] [CrossRef]
- Fiske, C.H.; Subbarow, Y. Determination of inorganic phosphate. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar]
- Kishino, A.; Yanagida, T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature 1988, 334, 74–76. [Google Scholar] [CrossRef]
- Borovikov, I.S.; Vinogradova, G.N.; Rozanov, I.M.; Barskii, I.I. Estimation of the magnitude of depolarization induced by microobjectives during polarization fluorescence measurements. Tsitologiia 1974, 16, 1045–1050. [Google Scholar]
- Tregear, R.T.; Mendelson, R.A. Polarization from a helix of fluorophores and its relation to that obtained from muscle. Biophys. J. 1975, 15, 455–467. [Google Scholar] [CrossRef]
- Kakol, I.; Borovikov, Y.S.; Szczesna, D.; Kirillina, V.P.; Levitsky, D.I. Conformational changes of F-actin in myosin-free ghost single fibre induced by either phosphorylated or dephosphorylated heavy meromyosin. Biochim. Biophys. Acta 1987, 913, 1–9. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation and Gene | Diagnosis | Ca2+-Sensitivity | Stiffness of Тpm | Shift of Тpm to the Inner Domain by TN | Shift of Тpm to the Inner Domain by S1 | Switching on of Actin by Tpm | Switching on of Actin by S1 | Strong Binding of S1 | Strong Binding of S1 in the Presence of ATP | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| High Ca2+ | Low Ca2+ | High Ca2+ | Low Ca2+ | High Ca2+ | Low Ca2+ | High Ca2+ | Low Ca2+ | High Ca2+ | Low Ca2+ | High Ca2+ | Low Ca2+ | ||||||
| E41K | TPM2 | NM [7,12] Cap [12] | ↓ | ↓ | ↓ | Norm | ↓ | ↑ | Norm | ↑ | ↓ | ↑ | ↓ | ↓ | ↑ | ↑ | [66] |
| R90P | TPM3 | CFTD [18] | ↑ | ↑ | ↑ | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↓ | ↑ | ↑ | ↓ | ↑ | Here |
| R91G | TPM2 | DA [69] | ↑ | ↓ | Norm | Norm | ↑ | ↑ | ↓ | ↓ | ↓ | ↑ | ↑ | ↑ | ↓ | ↑ | [64] |
| E117K | TPM2 | NM [70] CFTD [71] DA [7] | ↓ | ↑ | - | - | - | - | - | - | - | - | ↓ | ↓ | ↓ | ↓ | [67] |
| E139X | TPM2 | Cap [7,9,72] CFTD [7] | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | ↑ | ↓ | ↓ | ↑ | ↓ | ↑ | [5] |
| Q147P | TPM2 | Cap [71] NM [70] | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | ↓ | ↑ | ↓ | ↓ | ↓ | ↑ | ↓ | ↑ | [65] |
| E150A | TPM3 | Сap [7] | ↑ | ↓ | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | ↑ | Here |
| A155T | TPM3 | NM [19] | ↑ | ↓ | Norm | Norm | ↑ | ↑ | Norm | Norm | ↑ | ↑ | ↓ | ↑ | ↓ | ↑ | Here |
| A155T | TPM1 | - | - | ↓ | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | [57] |
| R167H | TPM1 | - | - | ↑ | ↓ | ↑ | ↓ | Norm | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | [51] |
| K168E | TPM1 | - | - | ↑ | Norm | Norm | ↓ | ↓ | ↓ | ↓ | Norm | ↓ | ↓ | ↑ | ↓ | ↑ | [51] |
| E173A | TPM3 | CFTD [17] | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | Here |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borovikov, Y.S.; Karpicheva, O.E.; Simonyan, A.O.; Avrova, S.V.; Rogozovets, E.A.; Sirenko, V.V.; Redwood, C.S. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. Int. J. Mol. Sci. 2018, 19, 3975. https://doi.org/10.3390/ijms19123975
Borovikov YS, Karpicheva OE, Simonyan AO, Avrova SV, Rogozovets EA, Sirenko VV, Redwood CS. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. International Journal of Molecular Sciences. 2018; 19(12):3975. https://doi.org/10.3390/ijms19123975
Chicago/Turabian StyleBorovikov, Yurii S., Olga E. Karpicheva, Armen O. Simonyan, Stanislava V. Avrova, Elena A. Rogozovets, Vladimir V. Sirenko, and Charles S. Redwood. 2018. "The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies" International Journal of Molecular Sciences 19, no. 12: 3975. https://doi.org/10.3390/ijms19123975
APA StyleBorovikov, Y. S., Karpicheva, O. E., Simonyan, A. O., Avrova, S. V., Rogozovets, E. A., Sirenko, V. V., & Redwood, C. S. (2018). The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. International Journal of Molecular Sciences, 19(12), 3975. https://doi.org/10.3390/ijms19123975