PI3Kinases in Diabetes Mellitus and Its Related Complications

Abstract
:1. Introduction
2. PI3K and Glucose Plasma Levels
3. PI3K and Diabetic Cardiomyopathy
4. PI3K and Diabetic Vasculopathy
5. PI3K and Diabetic Neuropathy and Encephalopathy
6. PI3K and Diabetic Nephropathy
7. Concluding Remarks
Funding
References
- World Health Organization (WHO). Global Report on Diabetes; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- King, G.L.; Park, K.; Li, Q. Selective Insulin Resistance and the Development of Cardiovascular Diseases in Diabetes: The 2015 Edwin Bierman Award Lecture. Diabetes 2016, 65, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, K.; Bernardo, B.C.; McMullen, J.R.; Ritchie, R.H. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther 2014, 142, 375–415. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Mensah, F.A.; Blaize, J.P.; Bryan, L.J. Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: Evidence to date. Onco Targets Ther. 2018, 11, 4817–4827. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Kapeller, R.; White, M.F.; Cantley, L.C. Activation of phosphatidylinositol 3-kinase by insulin. Proc. Natl. Acad. Sci. USA 1990, 87, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M.; Myers, M.G., Jr.; Shoelson, S.E.; Chin, D.J.; Sun, X.J.; Miralpeix, M.; Hu, P.; Margolis, B.; Skolnik, E.Y.; Schlessinger, J.; et al. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992, 11, 3469–3479. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.I.; Imai, Y.; Caruso, M.; Beguinot, F.; Formisano, P.; Accili, D. Evidence that IRS-2 phosphorylation is required for insulin action in hepatocytes. J. Biol. Chem. 1998, 273, 17491–17497. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Ito, K.; Todaka, M.; Hayashi, H.; Kamohara, S.; Ishii, K.; Okada, T.; Hazeki, O.; Ui, M.; Ebina, Y. Insulin-stimulated GLUT4 translocation is relevant to the phosphorylation of IRS-1 and the activity of PI3-kinase. Biochem. Biophys. Res. Commun. 1993, 195, 762–876. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Kawano, Y.; Sakakibara, T.; Hazeki, O.; Ui, M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J. Biol. Chem. 1994, 269, 3568–3573. [Google Scholar] [PubMed]
- Brachmann, S.M.; Ueki, K.; Engelman, J.A.; Kahn, R.C.; Cantley, L.C. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol. Cell. Biol. 2005, 25, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.; Ejeskär, K.; Wettergren, Y.; Kahn, C.R.; Rotter Sopasakis, V. Hepatic deletion of p110α and p85α results in insulin resistance despite sustained IRS1-associated phosphatidylinositol kinase activity. F1000Research 2017, 6, 1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, J.; Emmas, S.A.; Ble, F.X.; Barjat, H.; Lawrie, E.; Hancox, U.; Polanska, U.M.; Pritchard, A.; Hudson, K. The use of 18F-Fluoro-deoxy-glucose positron emission tomography (18F-FDG PET) as a non-invasive pharmacodynamic biomarker to determine the minimally pharmacologically active dose of AZD8835, a novel PI3Kα inhibitor. PLoS ONE 2017, 12, e0183048. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.; Emmas, S.A.; Blé, F.X.; Barjat, H.; Lawrie, E.; Hancox, U.; Oakes, D.; Polanska, U.M.; Barry, S.T. The use of (18)F-fluorodeoxyglucose positron emission tomography ((18)F-FDG PET) as a pathway-specific biomarker with AZD8186, a PI3Kβ/δ inhibitor. EJNMMI Res. 2016, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Hale, P.J.; López-Yunez, A.M.; Chen, J.Y. Genome-wide meta-analysis of genetic susceptible genes for Type 2 Diabetes. BMC Syst. Biol. 2012, 6 (Suppl. 3), S16. [Google Scholar] [CrossRef] [Green Version]
- Maffei, A.; Cifelli, G.; Carnevale, R.; Iacobucci, R.; Pallante, F.; Fardella, V.; Fardella, S.; Hirsch, E.; Lembo, G.; Carnevale, D. PI3Kγ Inhibition Protects Against Diabetic Cardiomyopathy in Mice. Rev. Esp. Cardiol. 2017, 70, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, N.; Mejia, E.M.; Sheikholeslami, K.; Sher, A.A.; Hou, S.; Hatch, G.M.; Marshall, A.J. TAPP Adaptors control B Cell metabolism by modulating the Phosphatidylinositol 3-Kinase signaling pathway: A novel regulatory circuit preventing autoimmunity. J. Immunol. 2018, 201, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Weaver, J.D.; Strauss, L.; Herbel, C.; Seth, P.; Boussiotis, V.A. Immunometabolic regulations mediated by coinhibitory receptors and their impact on T Cell immune responses. Front. Immunol. 2017, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- Frydrych, L.M.; Bian, G.; O’Lone, D.E.; Ward, P.A.; Delano, M.J. Obesity and type 2 diabetes mellitus drive immune dysfunction, infection development, and sepsis mortality. J. Leukoc. Biol. 2018, 104, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Rao, X.; Zhong, J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.L.; Chicanne, G.; Valet, C.; Hill, J.M.; Voshol, P.J.; et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hirshman, M.F.; Aschenbach, W.G.; Goodyear, L.J. Contraction regulation of Akt in rat skeletal muscle. J. Biol. Chem. 2002, 277, 11910–11917. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Fuentealba, C.; Contreras-Ferrat, A.E.; Altamirano, F.; Espinosa, A.; Li, Q.; Niu, W.; Lavandero, S.; Klip, A.; Jaimovich, E. Electrical stimuli release ATP to increase GLUT4 translocation and glucose uptake via PI3Kγ-Akt-AS160 in skeletal muscle cells. Diabetes 2013, 62, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, W.M.; Smith, S.B.; James, R.F.; Clifton, A.D.; Doza, Y.N.; Cohen, P.; Docherty, K. The p38/reactivating kinase mitogen-activated protein kinase cascade mediates the activation of the transcription factor insulin upstream factor 1 and insulin gene transcription by high glucose in pancreatic beta-cells. J. Biol. Chem. 1997, 272, 20936–20944. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.; Roduit, R.; Susin, S.; Prentki, M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia 1999, 42, 856–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolic, J.; Manning Fox, J.E.; Chepurny, O.G.; Spigelman, A.F.; Ferdaoussi, M.; Schwede, F.; Holz, G.G.; MacDonald, P.E. PI3 kinases p110α and PI3K-C2β negatively regulate cAMP via PDE3/8 to control insulin secretion in mouse and human islets. Mol. Metab. 2016, 5, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.H.; Lu, P.; Wang, Y.N.; Tian, C.G.; Huang, X.H.; Feng, Z.G.; Zhang, J.L.; Chang, H.Y. Role of PI3K p110β in the differentiation of human embryonic stem cells into islet-like cells. Biochem. Biophys. Res. Commun. 2017, 488, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Shin, H.M.; Jung, H.; Lee, E.; Kim, T.K.; Kim, T.N.; Kwon, M.J.; Lee, S.H.; Rhee, B.D.; Park, J.H. Comparison of pancreatic beta cells and alpha cells under hyperglycemia: Inverse coupling in pAkt-FoxO1. Diabetes Res. Clin. Pract. 2017, 131, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham Study. Am. J. Cardiol 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Sun, H.; Kerfant, B.G.; Zhao, D.; Trivieri, M.G.; Oudit, G.Y.; Penninger, J.M.; Backx, P.H. Insulin-like growth factor-1 and PTEN deletion enhance cardiac L-type Ca2+ currents via increased PI3Kalpha/PKB signaling. Circ. Res. 2006, 98, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Jiang, Y.P.; Wang, W.; Xu, X.H.; Mathias, R.T.; Entcheva, E.; Ballou, L.M.; Cohen, I.S.; Lin, R.Z. Loss of cardiac phosphoinositide 3-kinase p110 alpha results in contractile dysfunction. Circulation 2009, 120, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Jiang, Y.P.; Xu, X.H.; Ballou, L.M.; Cohen, I.S.; Lin, R.Z. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes 2007, 56, 2780–2789. [Google Scholar] [CrossRef] [PubMed]
- Prakoso, D.; De Blasio, M.J.; Qin, C.; Rosli, S.; Kiriazis, H.; Qian, H.; Du, X.J.; Weeks, K.L.; Gregorevic, P.; McMullen, J.R.; et al. Phosphoinositide 3-kinase (p110α) gene delivery limits diabetes-induced cardiac NADPH oxidase and cardiomyopathy in a mouse model with established diastolic dysfunction. Clin. Sci. 2017, 131, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Meoli, D.F.; Moslehi, J.; Roden, D.M. Inhibition of the α-Subunit of Phosphoinositide 3-Kinase in Heart Increases Late Sodium Current and Is Arrhythmogenic. J. Pharmacol. Exp. Ther. 2018, 365, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Vecchione, C.; Mascio, G.; Esposito, G.; Cifelli, G.; Martinello, K.; Landolfi, A.; Selvetella, G.; Grieco, P.; Damato, A.; et al. PI3Kγ inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc. Res. 2012, 93, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Creager, M.A.; Lüscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [PubMed]
- Vecchione, C.; Patrucco, E.; Marino, G.; Barberis, L.; Poulet, R.; Aretini, A.; Maffei, A.; Gentile, M.T.; Storto, M.; Azzolino, O.; et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kgamma. J. Exp. Med. 2005, 201, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Cardiovascular disease in type 2 diabetes mellitus. Arch. Intern. Med. 2003, 163, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, G.F.; Canobbio, I.; Torti, M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv. Biol. Regul. 2015, 59, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Guillermet-Guibert, J.; Chicanne, G.; Cabou, C.; Jandrot-Perrus, M.; Plantavid, M.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010, 115, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O.; et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Wågberg, F.; Andersson, M.; Skärby, T.; Gustafsson, D. Exploration of efficacy and bleeding with combined phosphoinositide 3-kinase β inhibition and aspirin in man. J. Thromb. Haemost. 2015, 13, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, T.A.; Moore, S.F.; Williams, C.M.; Poole, A.W.; Vanhaesebroeck, B.; Hers, I. Phosphoinositide 3-kinases p110α and p110β have differential roles in insulin-like growth factor-1-mediated Akt phosphorylation and platelet priming. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Terrisse, A.D.; Laurent, P.A.; Garcia, C.; Gratacap, M.P.; Vanhaesebroeck, B.; Sié, P.; Payrastre, B. The class I phosphoinositide 3-kinases α and β control antiphospholipid antibodies-induced platelet activation. Thromb. Haemost. 2016, 115, 1138–1146. [Google Scholar] [PubMed] [Green Version]
- Fougerat, A.; Gayral, S.; Gourdy, P.; Schambourg, A.; Rückle, T.; Schwarz, M.K.; Rommel, C.; Hirsch, E.; Arnal, J.F.; Salles, J.P.; et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation 2008, 117, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Bosco, O.; Tropel, P.; Laffargue, M.; Calvez, R.; Altruda, F.; Wymann, M.; Montrucchio, G. Resistance to thromboembolism in PI3Kgamma-deficient mice. FASEB J. 2001, 15, 2019–2021. [Google Scholar] [CrossRef] [PubMed]
- Valet, C.; Levade, M.; Chicanne, G.; Bilanges, B.; Cabou, C.; Viaud, J.; Gratacap, M.P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B.; et al. A dual role for the class III PI3K, Vps34, in platelet production and thrombus growth. Blood 2017, 130, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, M.; Luo, D.; Yue, M.; Wang, S.; Chen, X.; Zhou, Y.; Wang, Y.; Cai, Y.; Hu, X.; et al. Class III PI3K positively regulates platelet activation and thrombosis via PI(3)P-directed function of NADPH Oxidase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Helke, C.J. Abnormal PI3 kinase/Akt signal pathway in vagal afferent neurons and vagus nerve of streptozotocin-diabetic rats. Mol. Brain Res. 2003, 110, 234–244. [Google Scholar] [CrossRef]
- Di, G.; Zhao, X.; Qi, X.; Zhang, S.; Feng, L.; Shi, W.; Zhou, Q. VEGF-B promotes recovery of corneal innervations and trophic functions in diabetic mice. Sci. Rep. 2017, 7, 40582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Liu, S. Electroacupuncture with high frequency at acupoint ST-36 induces regeneration of lost enteric neurons in diabetic rats via GDNF and PI3K/AKT signal pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R109–R118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, G.; He, F.; Zhang, L.; Yang, K.; Yu, H.; Zhou, J.; Gan, H. MicroRNA 375 modulates hyperglycemia-induced enteric glial cell apoptosis and diabetes-induced gastrointestinal dysfunction by targeting Pdk1 and repressing PI3K/Akt pathway. Sci. Rep. 2018, 8, 12681. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Dysregulation of PI3K-Akt-mTOR pathway in brain of streptozotocin-induced type 2 diabetes mellitus in Wistar rats. Lipids Health Dis. 2018, 17, 168. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.C.; Magalhães, N.S.; e Silva, P.M.; Martins, M.A.; Carvalho, V.F. Activation of PPAR-γ reduces HPA axis activity in diabetic rats by up-regulating PI3K expression. Exp. Mol. Pathol. 2016, 101, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol 2011, 225, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, L. Calycosin ameliorates diabetes-induced cognitive impairments in rats by reducing oxidative stress via the PI3K/Akt/GSK-3β signaling pathway. Biochem. Biophys. Res. Commun. 2016, 473, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Gan, G.S.; Liu, Y.M.; Xiao, J.S.; Liu, H.X.; Mei, B.; Zhang, J.J. Adiponectin attenuates streptozotocin-induced Tau hyperphosphorylation and cognitive deficits by rescuing PI3K/Akt/GSK-3β Pathway. Neurochem. Res. 2018, 43, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Pan, T.L.; Hsu, J.W.; Huang, K.L.; Su, T.P.; Li, C.T.; Lin, W.C.; Tsai, S.J.; Chang, W.H.; Chen, T.J.; et al. Risk of type 2 diabetes in adolescents and young adults with attention-deficit/hyperactivity disorder: A nationwide longitudinal study. J. Clin. Psychiatry 2018, 79. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Chen, T.; Sundquist, J.; Sundquist, K. Type 1 diabetes in parents and risk of attention deficit/hyperactivity disorder in offspring: A population-based study in Sweden. Diabetes Care 2018, 41, 770–774. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, I.; Fardella, V.; Fardella, S.; Pallante, F.; Ghigo, A.; Iacobucci, R.; Maffei, A.; Hirsch, E.; Lembo, G.; Carnevale, D. Lack of kinase-independent activity of PI3Kγ in locus coeruleus induces ADHD symptoms through increased CREB signaling. EMBO Mol. Med. 2015, 7, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Chen, S.; Ziyadeh, F.N. From the periphery of the glomerular capillary wall toward the center of disease: Podocyte injury comes of age in diabetic nephropathy. Diabetes 2005, 54, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Welsh, G.I.; Hale, L.J.; Eremina, V.; Jeansson, M.; Maezawa, Y.; Lennon, R.; Pons, D.A.; Owen, R.J.; Satchell, S.C.; Miles, M.J.; et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Carranza, A.; Musolino, P.L.; Villar, M.; Nowicki, S. Signaling cascade of insulin-induced stimulation of L-dopa uptake in renal proximal tubule cells. Am. J. Physiol.-Cell Physiol. 2008, 295, C1602–C1609. [Google Scholar] [CrossRef] [PubMed]
- Hookham, M.B.; O’Donovan, H.C.; Church, R.H.; Mercier-Zuber, A.; Luzi, L.; Curran, S.P.; Carew, R.M.; Droguett, A.; Mezzano, S.; Schubert, M.; et al. Insulin receptor substrate-2 is expressed in kidney epithelium and up-regulated in diabetic nephropathy. FEBS J. 2013, 280, 3232–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, H.; Jiang, M.; Zhao, J.; Fan, C.; Wang, Y.; Peng, W. Huangqi (astragalus) decoction ameliorates diabetic nephropathy via IRS1-PI3K-GLUT signaling pathway. Am. J. Transl. Res. 2018, 10, 2491–2501. [Google Scholar] [PubMed]
- Chen, F.; Sun, Z.; Zhu, X.; Ma, Y. Astilbin inhibits high glucose-induced autophagy and apoptosis through the PI3K/Akt pathway in human proximal tubular epithelial cells. Biomed. Pharmacother. 2018, 106, 1175–1181. [Google Scholar] [CrossRef] [PubMed]

 Inhibition.
Inhibition.
Inhibition.
Inhibition. Inhibition.
Inhibition.

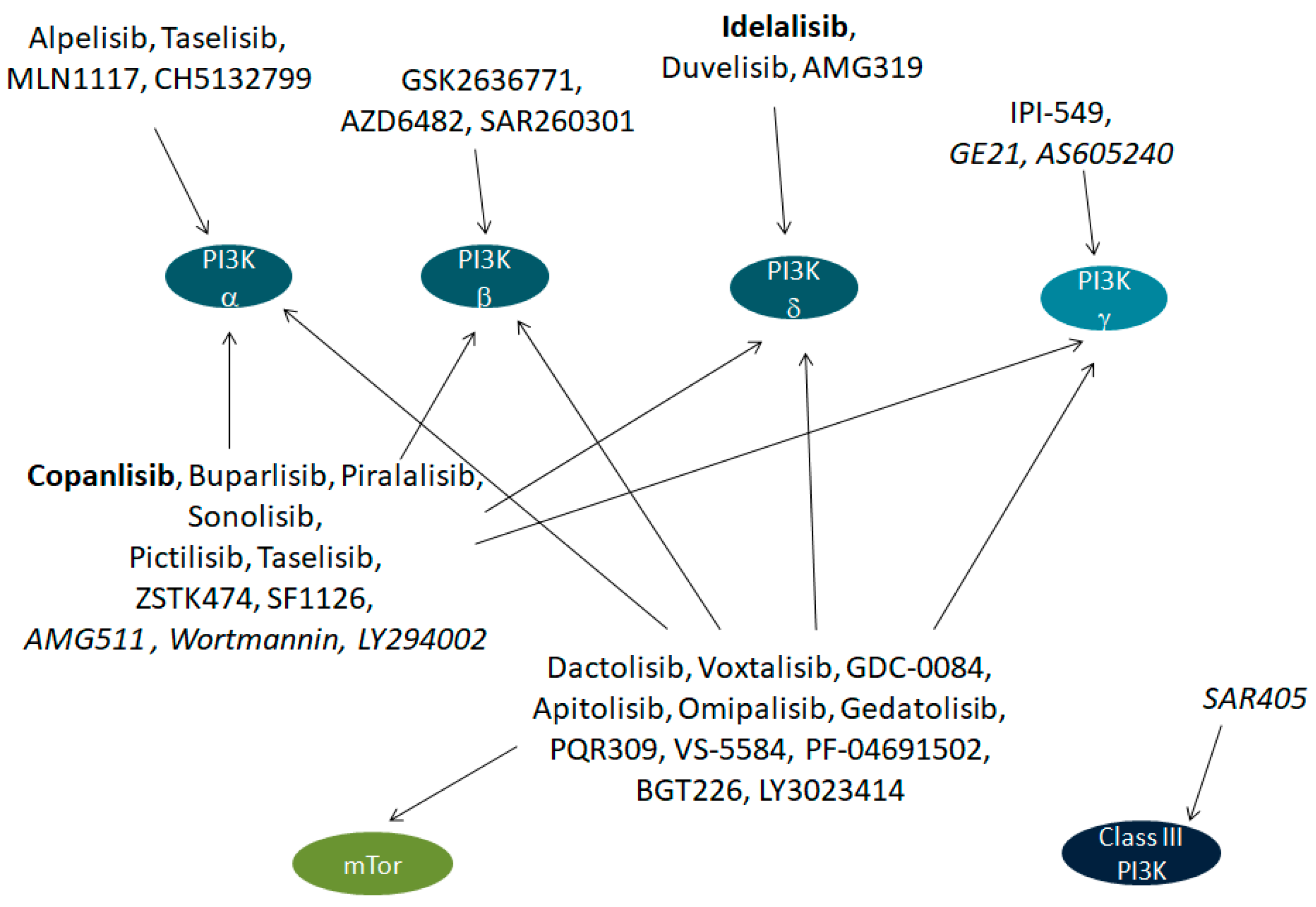{kind=link}
{kind=link}
{kind=link}
| Group | Isoform | Subunits | Main Activators | Main Substrate |
|---|---|---|---|---|
| Ia | PI3K α | p85 or p55 + p110 α | Tyrosine Kinase Receptors | Phosphatidylinositol 4,5-biphosphate |
| PI3K β | p85 or p55 + p110 β | |||
| PI3K δ | p85 or p55 + p110 δ | |||
| Ib | PI3K γ | p84/7 or p101 + p110 γ | G-Protein Coupled Receptors | |
| II | PI3K-C2α | Monomers | Tyrosine Kinase Receptors Or Cytokine receptors | Phosphatidylinositol or Phosphatidylinositol-4-phosphate |
| PI3K-C2β | ||||
| PI3K-C2γ | ||||
| III | PI3K-C3 | Vps34 + Vps15 | Rab | Phosphatidylinositol |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maffei, A.; Lembo, G.; Carnevale, D. PI3Kinases in Diabetes Mellitus and Its Related Complications. Int. J. Mol. Sci. 2018, 19, 4098. https://doi.org/10.3390/ijms19124098
Maffei A, Lembo G, Carnevale D. PI3Kinases in Diabetes Mellitus and Its Related Complications. International Journal of Molecular Sciences. 2018; 19(12):4098. https://doi.org/10.3390/ijms19124098
Chicago/Turabian StyleMaffei, Angelo, Giuseppe Lembo, and Daniela Carnevale. 2018. "PI3Kinases in Diabetes Mellitus and Its Related Complications" International Journal of Molecular Sciences 19, no. 12: 4098. https://doi.org/10.3390/ijms19124098
APA StyleMaffei, A., Lembo, G., & Carnevale, D. (2018). PI3Kinases in Diabetes Mellitus and Its Related Complications. International Journal of Molecular Sciences, 19(12), 4098. https://doi.org/10.3390/ijms19124098