Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Key Events in HCC
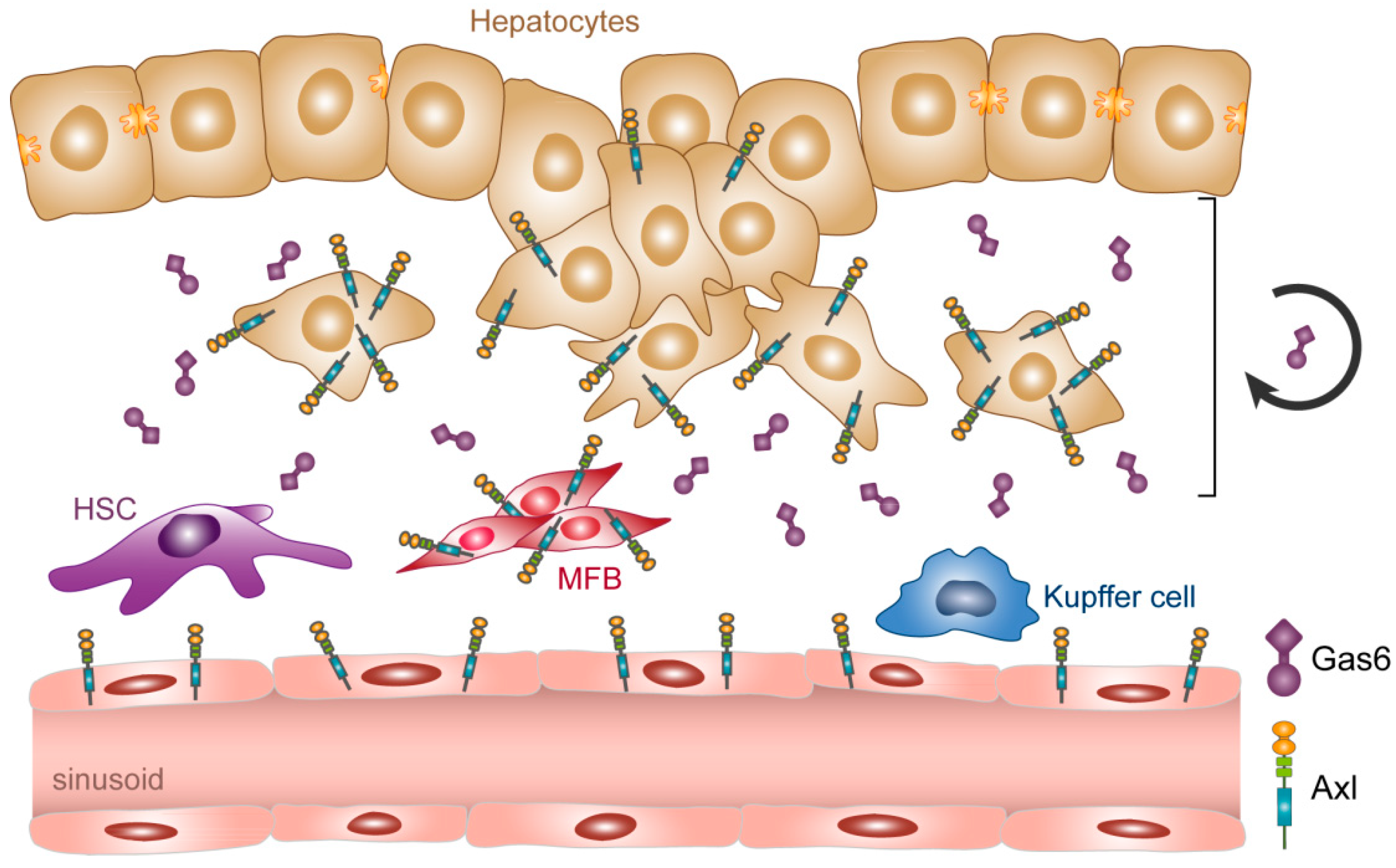
2. Biology of Gas6/Axl in the Liver
2.1. Receptor and Ligands
2.2. Dichotomic Role of Axl in Cancer
2.3. Role of Gas6/Axl under Healthy and Pathological Conditions in the Liver
3. Cell Communication Regulated by Ectodomain Shedding
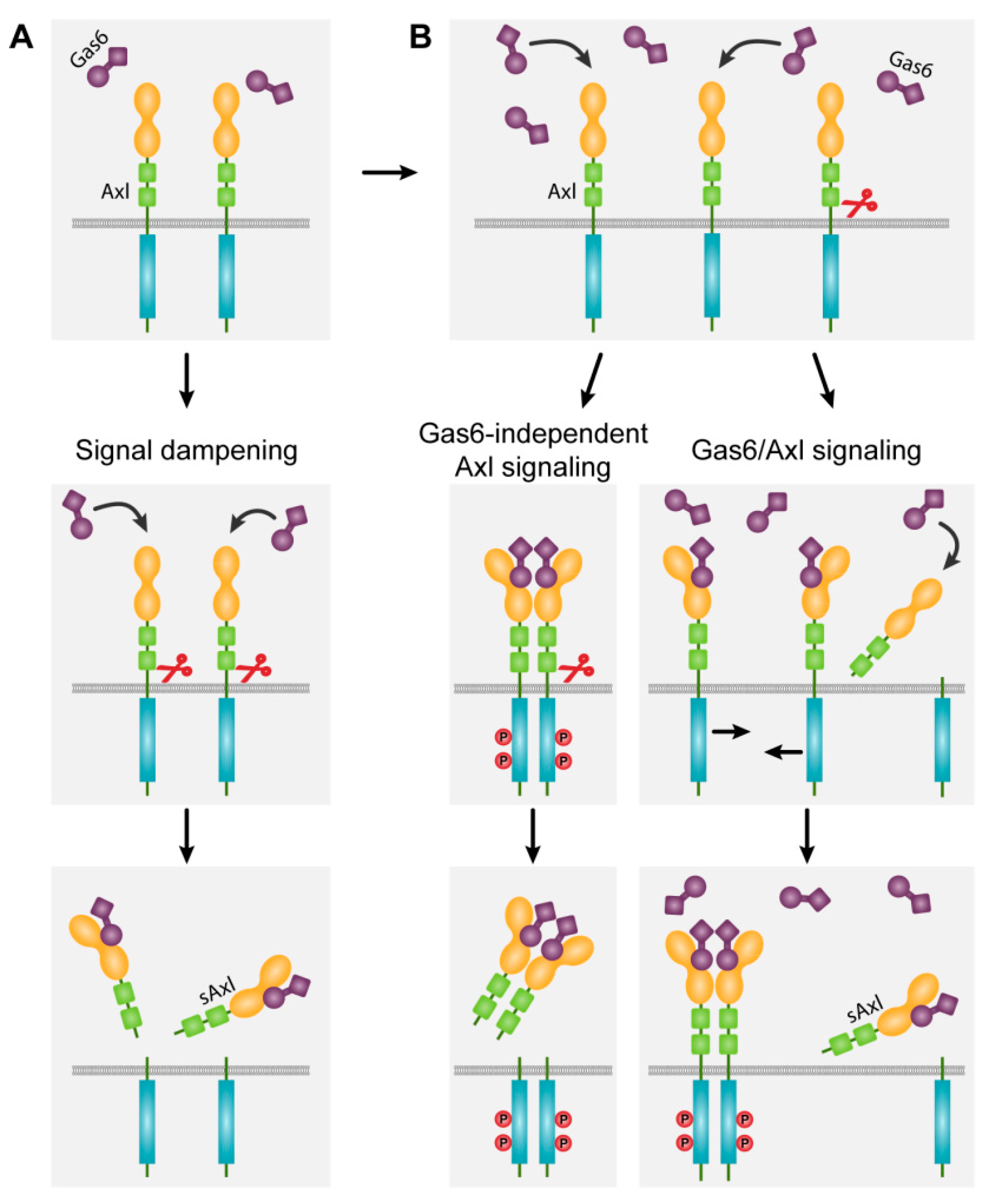
4. Axl Receptor Shedding: Gas6-Dependent Signaling versus Signal Dampening
5. Theranostics: Diagnostic and Therapeutic Potential of Gas6/Axl
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloproteinase |
| AFP | α-fetoprotein |
| BCLC | Barcelona Clinic Liver Cancer |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial to mesenchymal transition |
| ERK | extracellular-signal regulated kinase |
| FLT3 | FMS-like tyrosine kinase |
| Gas6 | growth arrest specific gene 6 |
| GVHD | graft versus host disease |
| HB-EGF | heparin-binding EGF-like growth factor |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| HER | human epidermal growth factor receptor |
| HIF | hypoxia- inducible factor |
| HSC | hepatic stellate cell |
| I/R | ischemia/reperfusion |
| ICD | intracellular domain |
| IFN | interferon |
| IFNAR | interferon α/β receptor |
| IL | interleukin |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| JNK | c-Jun-N-terminal kinase |
| KO | knock-out |
| MAPK | mitogen activated protein kinase |
| MAPKi | inhibition of the MAPK pathway |
| MCP | monocyte chemotactic protein |
| MMP | matrix metalloprotease |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NF-κB | nuclear factor κ-light-chain-enhancer of activated B cells |
| NK | natural killer |
| PDGFR | platelet-derived growth factor receptor |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| ProS | Protein S |
| RTK | receptor tyrosine kinase |
| sAxl | soluble Axl |
| sIL-6R | soluble interleukin-6 receptor |
| SOCS | suppressors of cytokine signaling |
| TAM | Tyro3, Axl, Mer |
| TGF | transforming growth factor |
| TKi | tyrosine kinase inhibitors |
| TLR | Toll-like receptor |
| TME | tumor microenvironment |
| TNF | tumor necrosis factor |
| VEGFR | vascular endothelial growth factor receptor |
| YAP | Yes-associated protein |
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Association for the Study of the Liver; European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Pressiani, T.; Personeni, N.; Santoro, A. Regorafenib for the treatment of unresectable hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2017, 17, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Zucman-Rossi, J. TERT promoter mutations in primary liver tumors. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.X.; Zhu, Q.G.; Zhang, S.M.; Guan, L.; Li, T.; Zhang, L.; Wang, S.Y.; Ren, W.L.; Chen, X.M.; Zhao, J.; et al. Precision medicine for hepatocellular carcinoma: Driver mutations and targeted therapy. Oncotarget 2017, 8, 55715–55730. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Villa, E.; Lahn, M. Transforming growth factor-β as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014, 74, 1890–1894. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.S.; Niu, X.J.; Wang, W.H. Genetic alterations in hepatocellular carcinoma: An update. World J. Gastroenterol. 2016, 22, 9069–9095. [Google Scholar] [CrossRef] [PubMed]
- Tsou, A.P.; Wu, K.M.; Tsen, T.Y.; Chi, C.W.; Chiu, J.H.; Lui, W.Y.; Hu, C.P.; Chang, C.; Chou, C.K.; Tsai, S.F. Parallel hybridization analysis of multiple protein kinase genes: Identification of gene expression patterns characteristic of human hepatocellular carcinoma. Genomics 1998, 50, 331–340. [Google Scholar] [CrossRef]
- Lanaya, H.; Natarajan, A.; Komposch, K.; Li, L.; Amberg, N.; Chen, L.; Wculek, S.K.; Hammer, M.; Zenz, R.; Peck-Radosavljevic, M.; et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat. Cell Biol. 2014, 16, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.H.; Hao, Y.L.; Zhu, J.W. Expression and prognostic value of HER-2/neu, STAT3 and SOCS3 in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gauglhofer, C.; Sagmeister, S.; Schrottmaier, W.; Fischer, C.; Rodgarkia-Dara, C.; Mohr, T.; Stattner, S.; Bichler, C.; Kandioler, D.; Wrba, F.; et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology 2011, 53, 854–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouissou, S.; La Bella, T.; Rekik, S.; Imbeaud, S.; Calatayud, A.L.; Rohr-Udilova, N.; Martin, Y.; Couchy, G.; Bioulac-Sage, P.; Grasl-Kraupp, B.; et al. Proliferation markers are associated with MET expression in hepatocellular carcinoma and predict tivantinib sensitivity in vitro. Clin. Cancer Res. 2017, 23, 4364–4375. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vilas, J.A.; Medina, M.A. Updates on the hepatocyte growth factor/c-Met axis in hepatocellular carcinoma and its therapeutic implications. World J. Gastroenterol. 2018, 24, 3695–3708. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, J.; Wang, X.; Shen, Q.; Li, C.; Dai, C. Co-expression of PDGF-B and VEGFR-3 strongly correlates with poor prognosis in hepatocellular carcinoma patients after hepatectomy. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhu, Y.; Han, L.; Ren, W.; Liu, H.; Qin, C. VEGFR-1 activation-induced MMP-9-dependent invasion in hepatocellular carcinoma. Future Oncol. 2015, 11, 3143–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.H.; Kim, J.M.; Kim, J.K.; Choi, S.J.; Lee, K.S.; Lee, J.W.; Chang, H.Y.; Lee, J.I. Platelet-derived growth factor receptor α in hepatocellular carcinoma is a prognostic marker independent of underlying liver cirrhosis. Oncotarget 2017, 8, 39534–39546. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wong, W.; Chua, S.C.; Wee, H.L.; Lim, S.G.; Chua, B.T.; Ho, H.K. Overexpression of Tyro3 and its implications on hepatocellular carcinoma progression. Int. J. Oncol. 2016, 48, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jia, L.; Ma, H.; Li, Y.; Ma, Z.; Zhao, Y. Axl gene knockdown inhibits the metastasis properties of hepatocellular carcinoma via PI3K/Akt-PAK1 signal pathway. Tumour Biol. 2014, 35, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Fuhrlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-β signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed]
- Heiring, C.; Dahlback, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Gohring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Varnum, B.C.; Young, C.; Elliott, G.; Garcia, A.; Bartley, T.D.; Fridell, Y.W.; Hunt, R.W.; Trail, G.; Clogston, C.; Toso, R.J.; et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 1995, 373, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Penninger, J.M. The role of TAM family receptors in immune cell function: Implications for cancer therapy. Cancers 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 8818–8852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [PubMed]
- Kirane, A.; Ludwig, K.F.; Sorrelle, N.; Haaland, G.; Sandal, T.; Ranaweera, R.; Toombs, J.E.; Wang, M.; Dineen, S.P.; Micklem, D.; et al. Warfarin blocks Gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 2015, 75, 3699–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Attar, E.C.; McCloskey, P.; Fridell, Y.W.; Liu, E.T. Determinants for transformation induced by the Axl receptor tyrosine kinase. Oncogene 1998, 16, 3177–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol. Cell. Biol. 1997, 17, 4442–4453. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Gas6-mediated survival in NIH3T3 cells activates stress signalling cascade and is independent of Ras. Oncogene 1999, 18, 4224–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, M.N.; Sosic, D.; Rothlin, C.V.; Kelly, E.; Lemke, G.; Olson, E.N.; Ivashkiv, L.B. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med. 2006, 203, 1891–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosic, D.; Richardson, J.A.; Yu, K.; Ornitz, D.M.; Olson, E.N. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-κB activity. Cell 2003, 112, 169–180. [Google Scholar] [CrossRef]
- Demarchi, F.; Verardo, R.; Varnum, B.; Brancolini, C.; Schneider, C. Gas6 anti-apoptotic signaling requires NF-κB activation. J. Biol. Chem. 2001, 276, 31738–31744. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S., Jr.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood 2007, 109, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Yttersian Sletta, K.; Tveitaras, M.K.; Lu, N.; Engelsen, A.S.T.; Reed, R.K.; Garmann-Johnsen, A.; Stuhr, L. Oxygen-dependent regulation of tumor growth and metastasis in human breast cancer xenografts. PLoS ONE 2017, 12, e0183254. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Jia, W.D.; Hu, B.; Pan, Y.Y. RAB10 overexpression promotes tumor growth and indicates poor prognosis of hepatocellular carcinoma. Oncotarget 2017, 8, 26434–26447. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, P.; Pierce, J.; Koski, R.A.; Varnum, B.; Liu, E.T. Activation of the Axl receptor tyrosine kinase induces mitogenesis and transformation in 32D cells. Cell. Growth Differ. 1994, 5, 1105–1117. [Google Scholar] [PubMed]
- O'Bryan, J.P.; Fridell, Y.W.; Koski, R.; Varnum, B.; Liu, E.T. The transforming receptor tyrosine kinase, Axl, is post-translationally regulated by proteolytic cleavage. J. Biol. Chem. 1995, 270, 551–557. [Google Scholar] [CrossRef] [PubMed]
- O'Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Orme, J.J.; Du, Y.; Vanarsa, K.; Mayeux, J.; Li, L.; Mutwally, A.; Arriens, C.; Min, S.; Hutcheson, J.; Davis, L.S.; et al. Heightened cleavage of Axl receptor tyrosine kinase by ADAM metalloproteases may contribute to disease pathogenesis in SLE. Clin. Immunol. 2016, 169, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekman, C.; Stenhoff, J.; Dahlback, B. Gas6 is complexed to the soluble tyrosine kinase receptor Axl in human blood. J. Thromb. Haemost. 2010, 8, 838–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merilahti, J.A.M.; Ojala, V.K.; Knittle, A.M.; Pulliainen, A.T.; Elenius, K. Genome-wide screen of γ-secretase-mediated intramembrane cleavage of receptor tyrosine kinases. Mol. Biol. Cell 2017, 28, 3123–3131. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular Pathways: Receptor Ectodomain Shedding in Treatment, Resistance, and Monitoring of Cancer. Clin. Cancer Res. 2017, 23, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loges, S.; Schmidt, T.; Tjwa, M.; van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jeng, Y.M.; Chen, Y.L.; Chung, L.; Yuan, R.H. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014, 35, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Sainaghi, P.P.; Castello, L.; Bergamasco, L.; Galletti, M.; Bellosta, P.; Avanzi, G.C. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. J. Cell. Physiol. 2005, 204, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, H.; Logsdon, C.D.; Rashid, A.; Fleming, J.B.; Abbruzzese, J.L.; Gomez, H.F.; Evans, D.B. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer 2011, 117, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [PubMed]
- Meric, F.; Lee, W.P.; Sahin, A.; Zhang, H.; Kung, H.J.; Hung, M.C. Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 2002, 8, 361–367. [Google Scholar] [PubMed]
- Antony, J.; Huang, R.Y. AXL-driven EMT state as a targetable conduit in cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Hafizi, S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017, 77, 2775–2778. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menendez, A.; Garcia-Ruiz, C.; Sancho-Bru, P.; Mari, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Valenti, L.; Marra, F.; Grimaudo, S.; Tripodo, C.; Bugianesi, E.; Camma, C.; Cappon, A.; Di Marco, V.; Di Maira, G.; et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Law, L.A.; Graham, D.K.; di Paola, J.; Branchford, B.R. GAS6/TAM pathway signaling in hemostasis and thrombosis. Front. Med. 2018, 5, 137. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Newman, P.J. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood 2004, 104, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; de Frutos, P.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.; Herbert, J.; et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, I.; Dittus, K.; Holmes, C.E. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013, 15, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Brummel-Ziedins, K.E.; Bouchard, B.A.; Holmes, C.E. Platelets in tumor progression: A host factor that offers multiple potential targets in the treatment of cancer. J. Cell. Physiol. 2014, 229, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Wen, J.Y.; Jia, C.C.; Wang, T.T.; Li, X.; Dong, M.; Lin, Q.U.; Chen, Z.H.; Ma, X.K.; Wei, L.I.; et al. MicroRNA-34a-5p enhances sensitivity to chemotherapy by targeting AXL in hepatocellular carcinoma MHCC-97L cells. Oncol. Lett. 2015, 10, 2691–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jia, L.; Liu, C.; Gong, Y.; Ren, D.; Wang, N.; Zhang, X.; Zhao, Y. Axl as a downstream effector of TGF-β1 via PI3K/Akt-PAK1 signaling pathway promotes tumor invasion and chemoresistance in breast carcinoma. Tumour Biol. 2015, 36, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Croucher, D.R.; Kennedy, S.P.; An, Q.; Pilkington, G.J.; Hafizi, S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis 2016, 5, e266. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The receptor tyrosine kinase AXL is required at multiple steps of the metastatic cascade during HER2-positive breast cancer progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Trahtemberg, U.; Mevorach, D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front. Immunol. 2017, 8, 1356. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.; Liu, P.; Zhang, Y.; Wu, H.; Chen, Y.; Han, D. Development of a spontaneous liver disease resembling autoimmune hepatitis in mice lacking tyro3, axl and mer receptor tyrosine kinases. PLoS ONE 2013, 8, e66604. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.K.; Wilhelm, A.; Antoniades, C.G. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G899–G905. [Google Scholar] [CrossRef] [PubMed]
- Murata, M. Inflammation and cancer. Environ. Health Prev. Med. 2018, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Kim, E.M.; Ji, K.Y.; Park, A.R.; Choi, H.R.; Lee, H.Y.; Kim, S.M.; Chung, B.Y.; Park, C.H.; Choi, H.J.; et al. Axl acts as a tumor suppressor by regulating LIGHT expression in T lymphoma. Oncotarget 2017, 8, 20645–20655. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Ben-Batalla, I.; Schultze, A.; Loges, S. Macrophage-tumor crosstalk: Role of TAMR tyrosine kinase receptors and of their ligands. Cell. Mol. Life Sci. 2012, 69, 1391–1414. [Google Scholar] [CrossRef] [PubMed]
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcot, A.; Couchie, D.; Chobert, M.N.; Zafrani, E.S.; Mavier, P.; Laperche, Y.; Brouillet, A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1043–G1053. [Google Scholar] [CrossRef] [PubMed]
- Lafdil, F.; Chobert, M.N.; Deveaux, V.; Zafrani, E.S.; Mavier, P.; Nakano, T.; Laperche, Y.; Brouillet, A. Growth arrest-specific protein 6 deficiency impairs liver tissue repair after acute toxic hepatitis in mice. J. Hepatol. 2009, 51, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnier, L.; Saller, F.; Kadi, L.; Brisset, A.C.; Sugamele, R.; Baudino, L.; Bono, F.; Herbert, J.-M.; Carmeliet, P.; Schapira, M.; et al. Gas6 deficiency in recipient mice of allogeneic transplantation alleviates hepatic graft-versus-host disease. Blood 2010, 115, 3390–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llacuna, L.; Barcena, C.; Bellido-Martin, L.; Fernandez, L.; Stefanovic, M.; Mari, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Garcia de Frutos, P.; Morales, A. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010, 52, 1371–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Yabkowitz, R.; Meyer, S.; Black, T.; Elliott, G.; Merewether, L.A.; Yamane, H.K. Inflammatory cytokines and vascular endothelial growth factor stimulate the release of soluble tie receptor from human endothelial cells via metalloprotease activation. Blood 1999, 93, 1969–1979. [Google Scholar] [PubMed]
- Wyble, C.W.; Hynes, K.L.; Kuchibhotla, J.; Marcus, B.C.; Hallahan, D.; Gewertz, B.L. TNF-α and IL-1 upregulate membrane-bound and soluble E-selectin through a common pathway. J. Surg. Res. 1997, 73, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Dummer, R.; Hartmann, A.A.; Burg, G.; Schmidt, R.E. Shedding of ICAM-1 from human melanoma cell lines induced by IFN-γ and tumor necrosis factor-α. Functional consequences on cell-mediated cytotoxicity. J. Immunol. 1991, 147, 4398–4401. [Google Scholar] [PubMed]
- Dethlefsen, S.M.; Raab, G.; Moses, M.A.; Adam, R.M.; Klagsbrun, M.; Freeman, M.R. Extracellular calcium influx stimulates metalloproteinase cleavage and secretion of heparin-binding EGF-like growth factor independently of protein kinase C. J. Cell. Biochem. 1998, 69, 143–153. [Google Scholar] [CrossRef]
- Arribas, J.; Massague, J. Transforming growth factor-α and β-amyloid precursor protein share a secretory mechanism. J. Cell Biol. 1995, 128, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.J.; Mason, J.C.; Lidington, E.A.; Edwards, D.R.; Nuttall, R.K.; Khokha, R.; Knauper, V.; Murphy, G.; Gavrilovic, J. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc. Res. 2005, 67, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walev, I.; Vollmer, P.; Palmer, M.; Bhakdi, S.; Rose-John, S. Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1996, 93, 7882–7887. [Google Scholar] [CrossRef] [PubMed]
- Borrell-Pages, M.; Rojo, F.; Albanell, J.; Baselga, J.; Arribas, J. TACE is required for the activation of the EGFR by TGF-α in tumors. EMBO J. 2003, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachmann, R.; Lindquist, P.B.; Nagashima, M.; Kohr, W.; Lipari, T.; Napier, M.; Derynck, R. Transmembrane TGF-α precursors activate EGF/TGF-α receptors. Cell 1989, 56, 691–700. [Google Scholar] [CrossRef]
- Horiuchi, K. A brief history of tumor necrosis factor α-converting enzyme: An overview of ectodomain shedding. Keio J. Med. 2013, 62, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tanida, S.; Joh, T.; Itoh, K.; Kataoka, H.; Sasaki, M.; Ohara, H.; Nakazawa, T.; Nomura, T.; Kinugasa, Y.; Ohmoto, H.; et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology 2004, 127, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, S.; Nanba, D. ADAM-mediated ectodomain shedding of HB-EGF in receptor cross-talk. Biochim. Biophys. Acta 2005, 1751, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Nanba, D.; Mammoto, A.; Hashimoto, K.; Higashiyama, S. Proteolytic release of the carboxy-terminal fragment of proHB-EGF causes nuclear export of PLZF. J. Cell Biol. 2003, 163, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, F.E. HER4 intracellular domain (4ICD) activity in the developing mammary gland and breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Linggi, B.; Cheng, Q.C.; Rao, A.R.; Carpenter, G. The ErbB-4 s80 intracellular domain is a constitutively active tyrosine kinase. Oncogene 2006, 25, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the AXL receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2017, 31, 1382–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foveau, B.; Ancot, F.; Leroy, C.; Petrelli, A.; Reiss, K.; Vingtdeux, V.; Giordano, S.; Fafeur, V.; Tulasne, D. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol. Biol. Cell 2009, 20, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Marron, M.B.; Singh, H.; Tahir, T.A.; Kavumkal, J.; Kim, H.Z.; Koh, G.Y.; Brindle, N.P. Regulated proteolytic processing of Tie1 modulates ligand responsiveness of the receptor-tyrosine kinase Tie2. J. Biol. Chem. 2007, 282, 30509–30517. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Wang, Z. HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance. Cancers 2017, 9. [Google Scholar] [CrossRef]
- Wang, J.; Willumsen, N.; Zheng, Q.; Xue, Y.; Karsdal, M.A.; Bay-Jensen, A.C. Bringing cancer serological diagnosis to a new level: Focusing on HER2, protein ectodomain shedding and neoepitope technology. Future Oncol. 2013, 9, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Segatto, O.; King, C.R.; Pierce, J.H.; Di Fiore, P.P.; Aaronson, S.A. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol. Cell. Biol. 1988, 8, 5570–5574. [Google Scholar] [CrossRef] [PubMed]
- Bellan, M.; Pogliani, G.; Marconi, C.; Minisini, R.; Franzosi, L.; Alciato, F.; Magri, A.; Avanzi, G.C.; Pirisi, M.; Sainaghi, P.P. Gas6 as a putative noninvasive biomarker of hepatic fibrosis. Biomark. Med. 2016, 10, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Fukuzawa, Y.; Matuyama, T.; Gotoh, K. Role of Tyro3, Axl, and Mer Receptors and Their Ligands (Gas6, and Protein S) in Patients with Hepatocellular Carcinoma. J. Cancer Ther. 2017, 8, 112–113. [Google Scholar] [CrossRef]
- Kohga, K.; Takehara, T.; Tatsumi, T.; Miyagi, T.; Ishida, H.; Ohkawa, K.; Kanto, T.; Hiramatsu, N.; Hayashi, N. Anticancer chemotherapy inhibits MHC class I-related chain a ectodomain shedding by downregulating ADAM10 expression in hepatocellular carcinoma. Cancer Res. 2009, 69, 8050–8057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, S.; Liu, K.; Wang, Y.; Ji, B.; Zhang, X.; Liu, Y. A disintegrin and metalloprotease (ADAM)10 is highly expressed in hepatocellular carcinoma and is associated with tumour progression. J. Int. Med. Res. 2014, 42, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichl, P.; Fang, M.; Starlinger, P.; Staufer, K.; Nenutil, R.; Muller, P.; Greplova, K.; Valik, D.; Dooley, S.; Brostjan, C.; et al. Multicenter analysis of soluble Axl reveals diagnostic value for very early stage hepatocellular carcinoma. Int. J. Cancer 2015, 137, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, M.; Staufer, K.; Huber, H.; Stauber, R.; Bantel, H.; Weiss, K.H.; Starlinger, P.; Pock, H.; Kloters-Plachky, P.; Gotthardt, D.N.; et al. Soluble Axl is an accurate biomarker of cirrhosis and hepatocellular carcinoma development: Results from a large scale multicenter analysis. Oncotarget 2017, 8, 46234–46248. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, M.B.; Bucher, J.N.; Vater, A.; Bazhin, A.V.; Hao, J.; Guba, M.O.; Angele, M.K.; Werner, J.; Rentsch, M. Resection or transplant in early hepatocellular carcinoma. Dtsch. Arztebl. Int. 2017, 114, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Batlle, M.; Recarte-Pelz, P.; Roig, E.; Castel, M.A.; Cardona, M.; Farrero, M.; Ortiz, J.T.; Campos, B.; Pulgarin, M.J.; Ramirez, J.; et al. AXL receptor tyrosine kinase is increased in patients with heart failure. Int. J. Cardiol. 2014, 173, 402–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Diep, A.; Nash, S.E.; Olcina, M.M.; Jiang, D.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Koong, A.C.; et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Investig. 2017, 127, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming growth factor-β and Axl induce CXCL5 and neutrophil recruitment in hepatocellular carcinoma. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 2016, 150, 1646–1658. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holstein, E.; Binder, M.; Mikulits, W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. Int. J. Mol. Sci. 2018, 19, 4111. https://doi.org/10.3390/ijms19124111
Holstein E, Binder M, Mikulits W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. International Journal of Molecular Sciences. 2018; 19(12):4111. https://doi.org/10.3390/ijms19124111
Chicago/Turabian StyleHolstein, Elisa, Mathias Binder, and Wolfgang Mikulits. 2018. "Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics" International Journal of Molecular Sciences 19, no. 12: 4111. https://doi.org/10.3390/ijms19124111
APA StyleHolstein, E., Binder, M., & Mikulits, W. (2018). Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. International Journal of Molecular Sciences, 19(12), 4111. https://doi.org/10.3390/ijms19124111