Apoptosis: A Target for Anticancer Therapy

Abstract
:1. Introduction
2. Apoptosis in Cancer
3. Apoptosis and Cancer Therapy
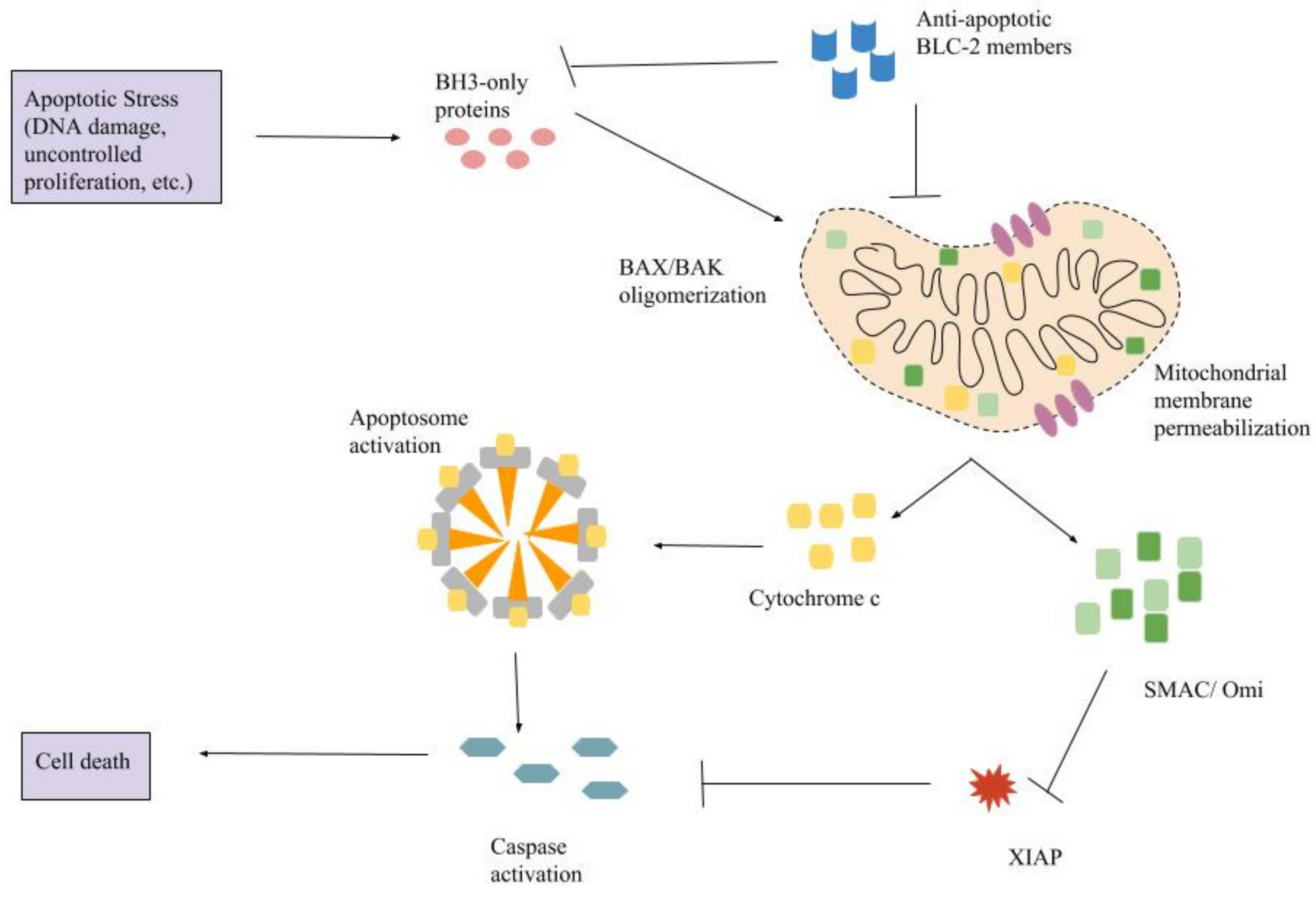
4. Intrinsic Pathway
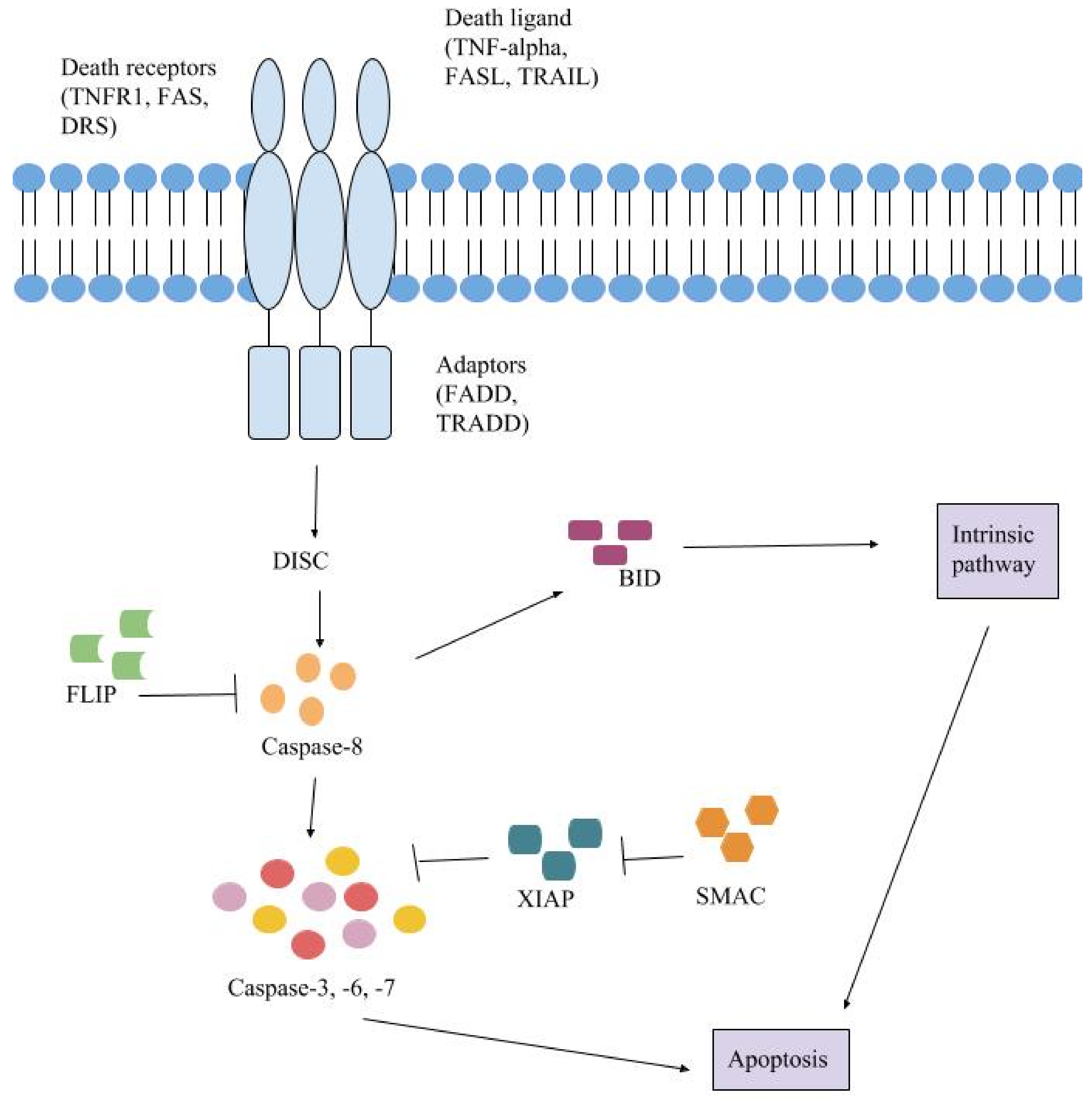
5. Extrinsic Pathway
6. Apoptotic Changes in Cancer
7. Proapoptotic Regulation in Tumor Cells
8. Prosurvival Regulation in Tumor Cells
9. Blebbishield Formation
10. Plant-Derived Compounds Exhibiting Anti-Cancerous Activity
11. Conclusions
Acknowledgments
Conflicts of Interest
References
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Zaman, S.; Wang, R.; Gandhi, V. Targeting the apoptosis pathway in hematologic malignancies. Leuk. Lymphoma 2014, 55, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Bonner, M.Y.; Gilbert, L.C. Targeting the duality of cancer. NPJ Precis. Oncol. 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Jing, L.; Wang, Q.; Lin, C.-C.; Chen, X.; Diao, J.; Liu, Y.; Sun, X. Bas-PGAM5L-Drp1 complex is required for intrinsic apoptosis execution. Oncotarget 2015, 6, 30017–30034. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative dieseaes. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Villa-Pulgarín, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marbán, A.; Müller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and lipid raft-located FoF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Zhang, Q.; Zhu, Z.; Xu, H.; Ding, F.; Wang, M.; Du, S.; Du, Y.; Yan, Z. BHX, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating Wnt/β-catenin signaling. Sci. Rep. 2017, 7, 9153. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27, S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradarn, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Su, D.; Zhang, J.; Ge, S.; Li, Y.; Wang, F.; Gravel, M.; Roulston, A.; Song, Q.; Xu, W.; et al. Improvement of pharmacokinetic profile of TRAIL via trimer-tage enhances its antitumor activity in vivo. Sci. Rep. 2017, 7, 8953. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Reed, J.C. BCL-2 family proteins and cancer. Oncogene 2008, 77, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Elkholi, R.; Renault, T.T.; Serasinghe, M.N.; Chipuk, J.E. Putting the pieces together: How is the mitochondrial pathway of apoptosis regulated in cancer and chemotherapy? Cancer Metab. 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Burmester, T.; Hankeln, T. What is the function of neuroglobin? J. Exp. Biol. 2009, 212, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Fiocchetti, M.; Nuzzo, M.T.; Totta, P.; Acconcia, F.; Ascenzi, P.; Marino, M. Neuroglobin, a pro-survival player in estrogen receptor α-positive cancer cells. Cell Death Dis. 2014, 5, e1449. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Kamat, A.M. Endocytosis and serpentine filopodia drive blebbishield-mediated resurrection of apoptotic cancer stem cells. Cell Death Discov. 2016, 2, 15069. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Choi, W.; Shah, J.B.; Lee, E.K.; Willis, D.L.; Kamat, A.M. Blebbishields, the emergency program for cancer stem cells: Sphere formation and tumorigenesis after apoptosis. Cell Death Differ. 2013, 20, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G. Exposing the deadly dark side of apoptotic cancer stem cells. Oncoscience 2017, 4, 124–125. [Google Scholar] [PubMed]
- Jinesh, G.G.; Molina, J.R.; Huang, L.; Laing, N.M.; Mills, G.B.; Var-Eli, M.; Kamat, A.M. Mitochondrial oligomers boost glycolysis in cancer stem cells to facilitate blebbishield-mediated transformation after apoptosis. Cell Death Discov. 2016, 2, 16003. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Kamat, A.M. Blebbishield emergency program: An apoptotic route to cellular transformation. Cell Death Differ. 2016, 23, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, M.; Kapulnik, Y.; Koltai, H. Plant derived substances with anti-cancer activity: From folklore to practice. Front. Plant Sci. 2015, 6, 799. [Google Scholar] [CrossRef] [PubMed]
- Ioannis, P.; Anastasis, S.; Andreas, Y. Graviola: A systematic review on its anticancer properties. Am. J. Cancer Prev. 2015, 3, 128–131. [Google Scholar]
- Yu-Min, K.; Tung-Ying, W.; Yang-Chang, W.; Fang-Rong, C.; Jinn-Yuh, G.; Lea-Yea, C. Annonacin induces cell cycle-dependent growth arrest and apoptosis in estrogen receptor-α-related pathways in MCF-7 cells. J. Ethnopharmacol. 2011, 137, 1283–1290. [Google Scholar]
- Grant, P.; Ramasamy, S. An update on plant derived anti-androgens. Int. J. Endocrinol. Metab. 2012, 10, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Bao, J.L.; Wu, G.S.; Xu, W.S.; Huang, M.Q.; Chen, X.P.; Wang, Y.T. Quinones derived from plant secondary metabolites as anti-cancer agents. Anticancer Agents Med. Chem. 2013, 13, 456–463. [Google Scholar] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, D.O.; Dembitsky, V.M. Anti-breast cancer agents derived from plants. Nat. Prod. Bioprospect. 2014, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dalimi, A.; Delavari, M.; Ghaffarifar, F.; Sadraei, J. In vitro and in vivo antileishmanial effects of aloe-emodin on Leishmania major. J. Tradit. Complement. Med. 2015, 5, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, J.; Prasad, S.; Aggarwal, B.B. Curcumin and cancer cells: How many ways can curry kill tumor cells selectively? AAPS J. 2009, 11, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.I.; Braicu, C.; Zanoaga, O.; Pileczki, V.; Gherman, C.; Berindan-Neagoe, I.; Campian, R.S. Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis and autophagy in oral cancer SSC-4 cells. OncoTargets Ther. 2015, 8, 461–470. [Google Scholar]
- Zhang, Z.; Wang, C.-H.; Du, G.-J.; Qi, L.-W.; Calway, T.; He, T.-C.; Du, W.; Yuan, C.-S. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int. J. Oncol. 2013, 43, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, H.; Xu, Y.; Zhang, J.; Zhu, W.; Zhang, Y.; Mao, Y. Juglone induces apoptosis of tumor stem-like cells through ROS-p38 pathway in glioblastoma. BMC Neurol. 2017, 17, 70. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Halagowder, D.; Sivasithambaram, N.D. Quercetin suppresses twist to induce apoptosis in MCF-7 breast cancer cells. PLoS ONE 2015, 10, e0141370. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Nagesh, P.K.B.; Jaggi, M.; Chauhan, S.C. Therapeutic applications of curcumin nanoformulations. AAPS J. 2015, 17, 1341–1356. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.H.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M. Therapeutic applications of curcumin for patients with pancreatic cancer. World J. Castroenterol. 2014, 20, 9384–9391. [Google Scholar]
- Heger, M.; van Golen, R.F.; Broekgaarden, M.; Michel, M.C. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer. Pharmacol. Rev. 2014, 66, 222–307. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.T.K.; Ghosh, M.; Forte, T.M.; Ryan, R.O.; Gordon, L.I. Curcumin nanodisk induced apoptosis in mantle cell lymphoma. Leuk. Lymphoma 2011, 52, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Singh, A.T.K.; Xu, W.; Sulchek, T.; Gordon, L.I.; Ryan, R.O. Curcumin Nanodisks: Formulation and Characterization. Nanomedicine 2011, 7, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Crosby, N.M.; Ghosh, M.; Su, B.; Beckstead, J.A.; Kamei, A.; Simonsen, J.B.; Luo, B.; Gordon, L.I.; Forte, T.M.; Ryan, R.O. Anti-CD20 single chain variable antibody fragment–apolipoprotein A-I chimera containing nanodisks promote targeted bioactive agent delivery to CD20-positive lymphomas. Biochem. Cell Biol. 2015, 93, 1–8. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | Found in | Mechanism |
|---|---|---|
| Aloe-emodin | Rheum palmatum | Induces cytochrome c release [32] |
| Black cohosh | Actaea racemosa | Activates caspases [28] |
| Curcumin | Tumeric | Inhibits BCL-2 and XIAP [33] |
| Epigallocatechin-3-gallate | Green tea component | Activates cell death receptors [34] |
| Genistein | Soybeans | Cell cycle arrest activation [35] |
| Graviola | Annona muricata | Inhibits BCL-2 and activates BAX [26] |
| Juglone | Juglans mandshurica | Increase caspase 9 cleavage [36] |
| Quercetin | Bark of many plants | Modulating cell cycle regulators to arrest the cell cycle [37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. https://doi.org/10.3390/ijms19020448
Pfeffer CM, Singh ATK. Apoptosis: A Target for Anticancer Therapy. International Journal of Molecular Sciences. 2018; 19(2):448. https://doi.org/10.3390/ijms19020448
Chicago/Turabian StylePfeffer, Claire M., and Amareshwar T. K. Singh. 2018. "Apoptosis: A Target for Anticancer Therapy" International Journal of Molecular Sciences 19, no. 2: 448. https://doi.org/10.3390/ijms19020448
APA StylePfeffer, C. M., & Singh, A. T. K. (2018). Apoptosis: A Target for Anticancer Therapy. International Journal of Molecular Sciences, 19(2), 448. https://doi.org/10.3390/ijms19020448