Differential Inflammatory-Response Kinetics of Human Keratinocytes upon Cytosolic RNA- and DNA-Fragment Induction

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
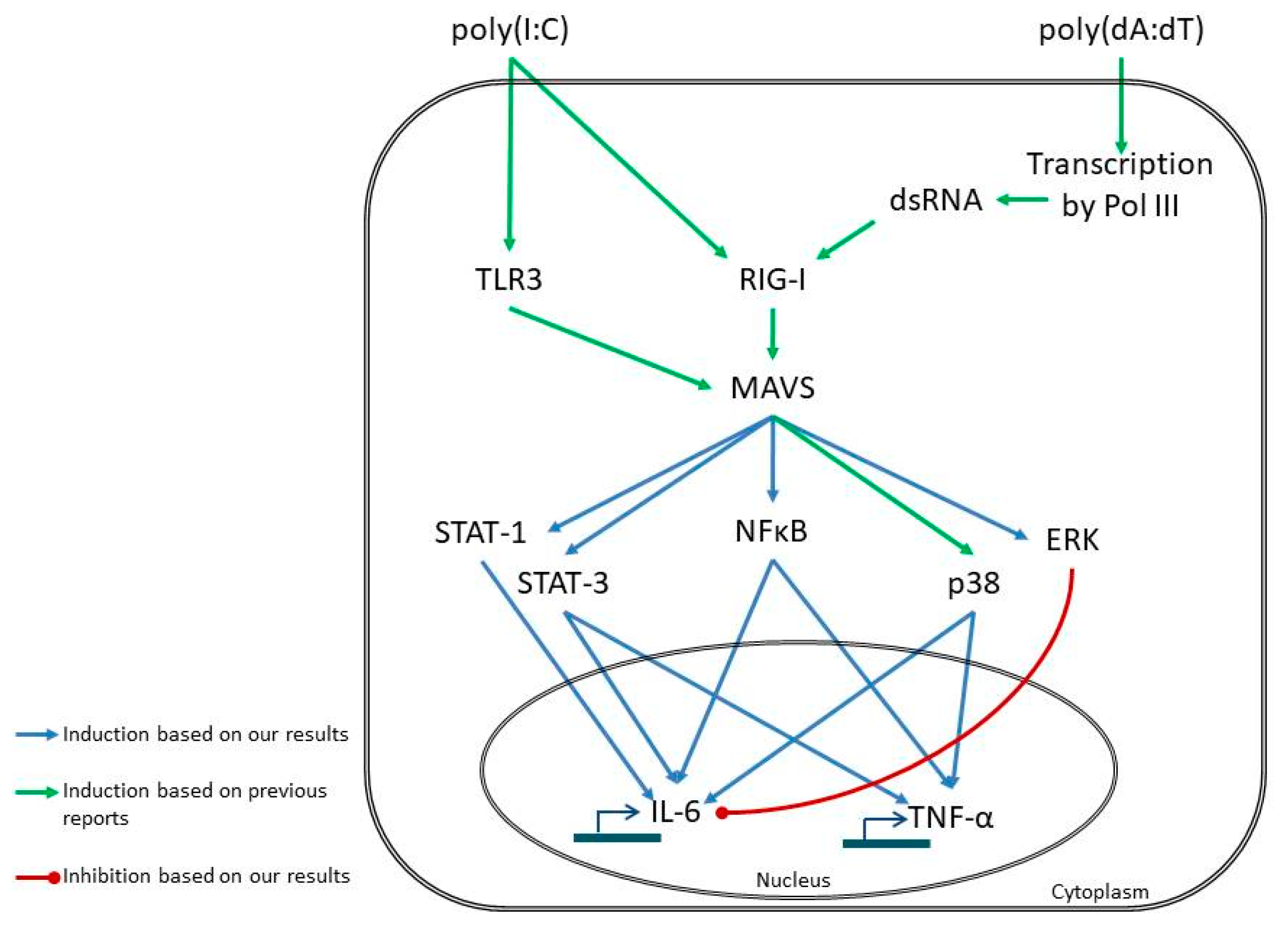{kind=link}
Abstract
:1. Introduction
2. Results
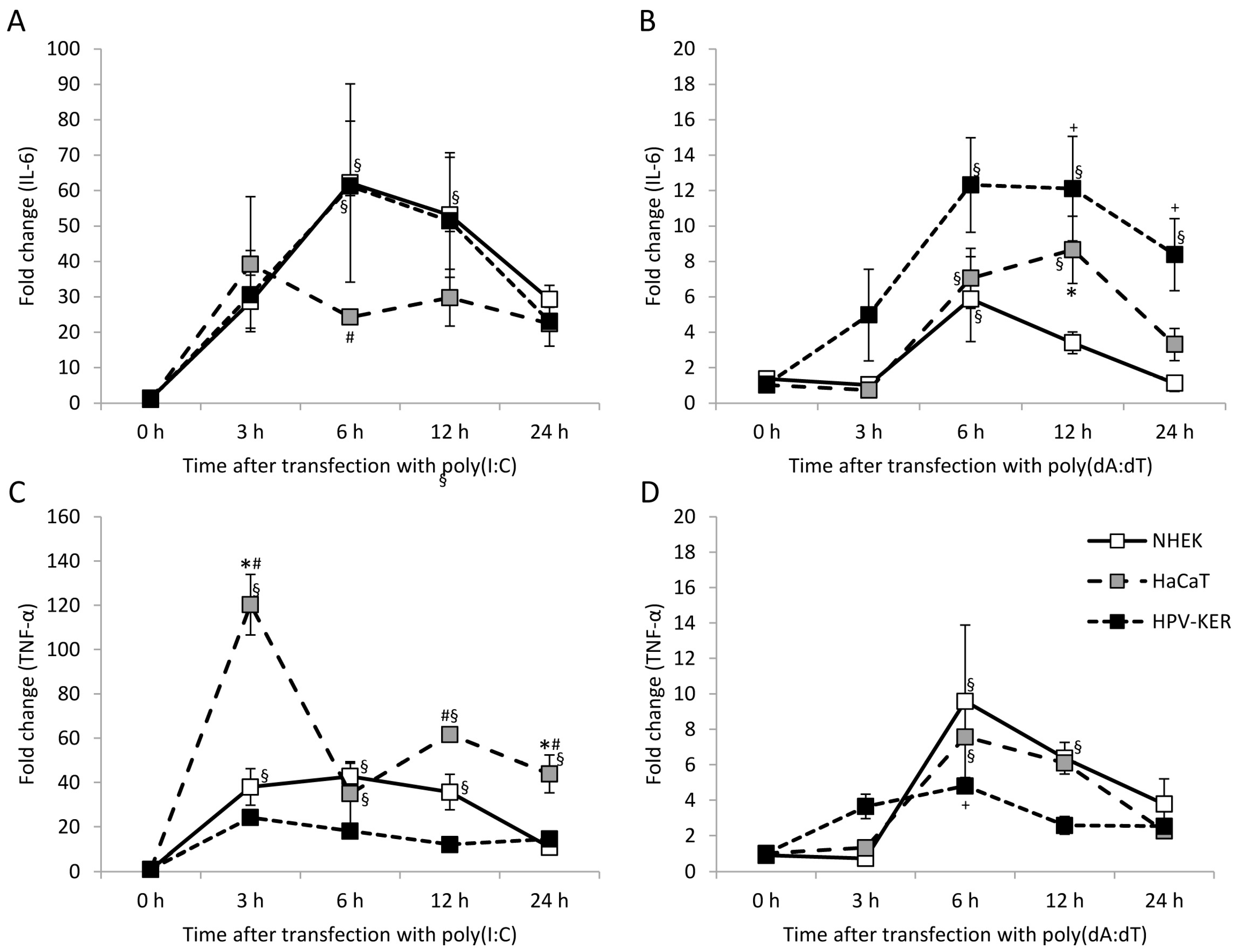
2.1. Keratinocytes Respond to Poly(I:C) and Poly(dA:dT) with Increased Interleukin-6 (IL-6) and Tumor Necrosis Factor α (TNF-α) Expression
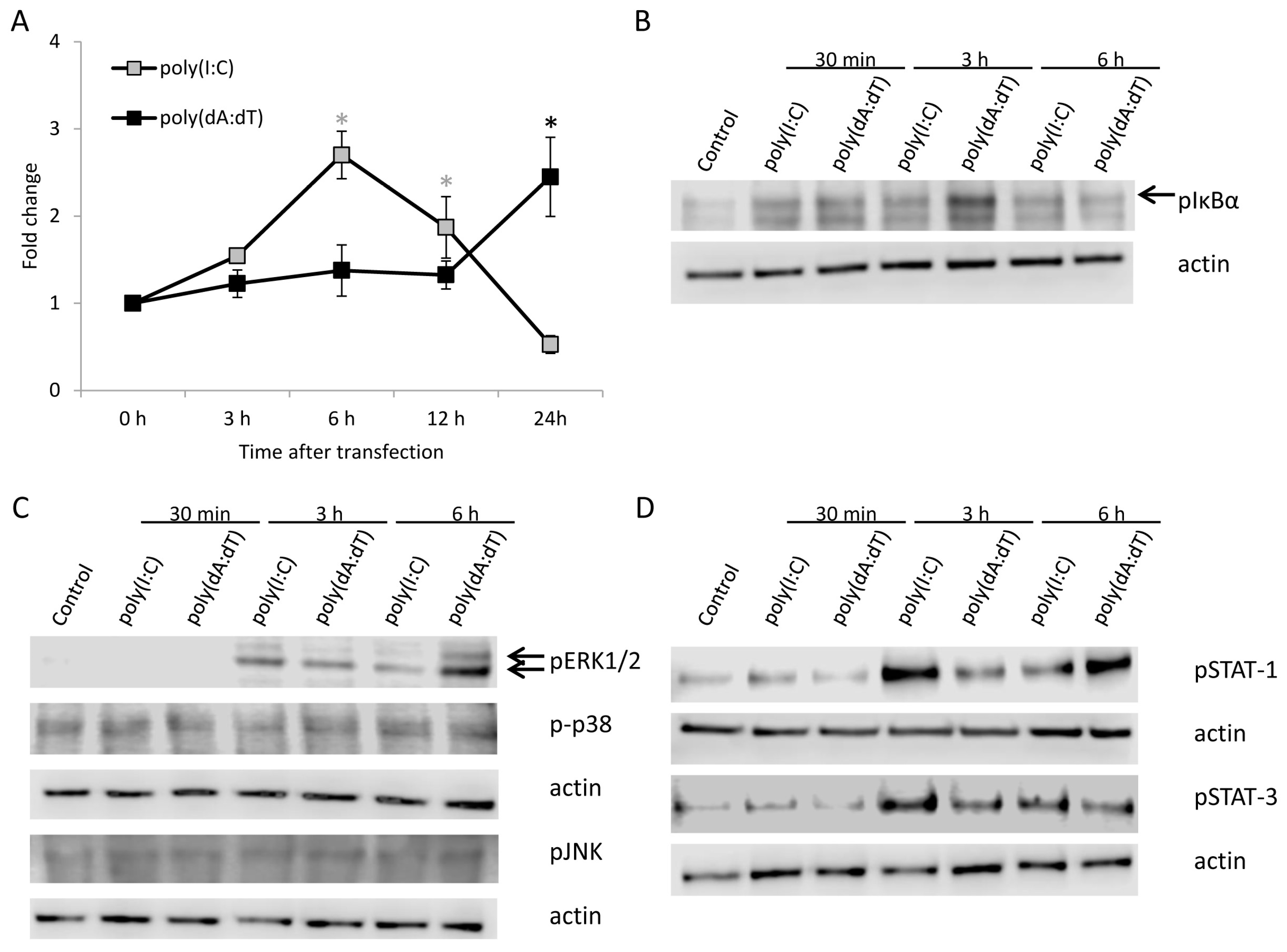
2.2. Poly(I:C) and Poly(dA:dT) Treatment Induces Nuclear Factor κB (NF-κB), Mitogen Activated Protein Kinase (MAPK) and Signal Transducers of Activator of Transcription (STAT) Activation in Keratinocytes
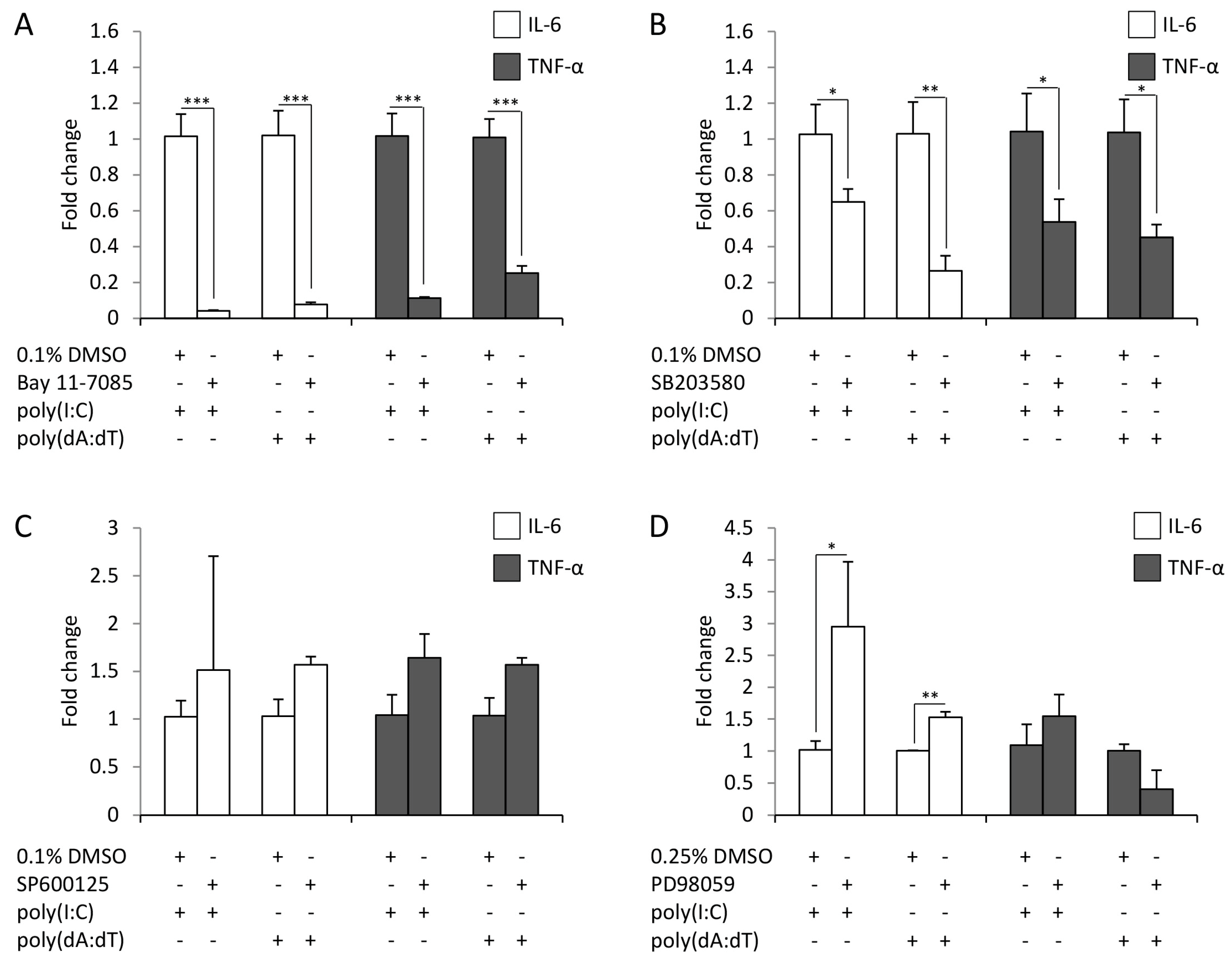
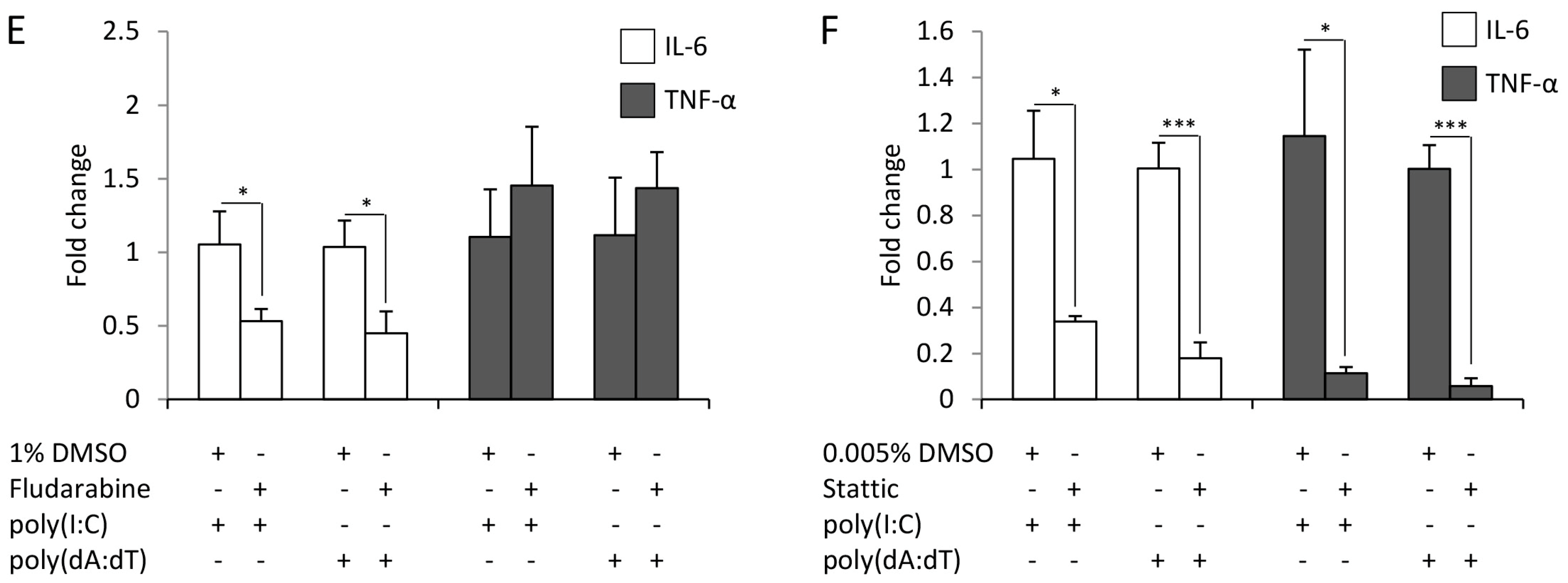
2.3. Cytokine Expression of Keratinocytes upon Poly(I:C) and Poly(dA:dT) Treatment Relies on NF-κB, p38 and STAT Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Stimulation of the Cells
4.3. RNA Isolation and RT-PCR
4.4. Detection of NF-κB Induction
4.5. Western Blot Analysis
4.6. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IL-6 | interleukin-6 |
| TNF-α | tumor necrosis factor α |
| PAMP | pathogen associated molecular pattern |
| DAMP | damage associated molecular pattern |
| DNase | deoxyribonuclease |
| AIM2 | absent in melanoma 2 |
| IFN | interferon |
| TLR | toll like receptor |
| RIG-I | retinoic acid induced gene I |
| cGAS | cyclic GMP-AMP synthase |
| ds | double-stranded |
| poly(dA:dT) | polydeoxyadenylic acid-polydeoxythymidylic acid double-stranded homopolymer |
| poly(I:C) | Polyinosinic-polycytidylic acid |
| MAPK | mitogen activated protein kinase |
| JNK | c-Jun N-terminal kinase |
| ERK1/2 | extracellular signal-regulated protein kinase 1 and 2 |
| MEK1/2 | dual specificity mitogen-activated protein kinase kinase 1 and 2 |
| NF-κB | nuclear factor κB |
| IκBα | NF-κB inhibitor α |
| STAT | signal transducer and activator of transcription |
| NHEK | normal human epidermal keratinocyte |
References
- Pivarcsi, A.; Koreck, A.; Bodai, L.; Széll, M.; Szeg, C.; Belso, N.; Kenderessy-Szabó, A.; Bata-Csörgo, Z.; Dobozy, A.; Kemény, L. Differentiation-regulated expression of toll-like receptors 2 and 4 in HaCat keratinocytes. Arch. Dermatol. Res. 2004, 296, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Köllisch, G.; Kalali, B.N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005, 114, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.S.; Ovigne, J.-M.; Powles, A.V.; Corcoran, S.; Fry, L. Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: Modulation of TLR expression in chronic plaque psoriasis. Br. J. Dermatol. 2003, 148, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Mempel, M.; Voelcker, V.; Köllisch, G.; Plank, C.; Rad, R.; Gerhard, M.; Schnopp, C.; Fraunberger, P.; Walli, A.K.; Ring, J.; et al. Toll-like receptor expression in human keratinocytes: Nuclear factor κB controlled gene activation by Staphylococcus aureus is toll-like receptor 2 but not toll-like receptor 4 or platelet activating factor receptor dependent. J. Investig. Dermatol. 2003, 121, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Luff, J.A.; Yuan, H.; Kennedy, D.; Schlegel, R.; Felsburg, P.; Moore, P.F. Keratinocyte antiviral response to poly(dA:dT) stimulation and papillomavirus infection in a canine model of X-linked severe combined immunodeficiency. PLoS ONE 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [PubMed]
- Prens, E.P.; Kant, M.; van Dijk, G.; van der Wel, L.I.; Mourits, S.; van der Fits, L. IFN-alpha enhances poly-IC responses in human keratinocytes by inducing expression of cytosolic innate RNA receptors: Relevance for psoriasis. J. Investig. Dermatol. 2008, 128, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Tervaniemi, M.; Katayama, S.; Skoog, T.; Siitonen, H.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S.; et al. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Shao, S.; Li, B.; Jin, L.; Lei, J.; Qiao, H.; Wang, G. Up-regulation of Interferon-inducible protein 16 contributes to psoriasis by modulating chemokine production in keratinocytes. Sci. Rep. 2016, 6, 25381. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göss, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Molès, J.-P.; Griez, A.; Guilhou, J.-J.; Girard, C.; Nagot, N.; Van de Perre, P.; Dujols, P. Cytosolic RNA:DNA duplexes generated by endogenous reverse transcriptase activity as autonomous inducers of skin inflammation in psoriasis. PLoS ONE 2017, 12, e0169879. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Szabo, S.; Scherz, J.; Jaeger, K.; Rossiter, H.; Buchberger, M.; Ghannadan, M.; Hermann, M.; Theussl, H.-C.; Tobin, D.J.; et al. essential role of the keratinocyte-specific endonuclease DNase1L2 in the removal of nuclear DNA from hair and nails. J. Investig. Dermatol. 2011, 131, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Buchberger, M.; Napirei, M.; Tschachler, E.; Eckhart, L. Inactivation of DNase1L2 and DNase2 in keratinocytes suppresses DNA degradation during epidermal cornification and results in constitutive parakeratosis. Sci. Rep. 2017, 7, 6433. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Romeu, E.; Ferran, M.; Giménez-Arnau, A.; Bugara, B.; Lipert, B.; Jura, J.; Florencia, E.F.; Prens, E.P.; Celada, A.; Pujol, R.M.; et al. MCPIP1 RNase is aberrantly distributed in psoriatic epidermis and rapidly induced by IL-17A. J. Investig. Dermatol. 2016, 136, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Kopfnagel, V.; Wagenknecht, S.; Harder, J.; Hofmann, K.; Kleine, M.; Buch, A.; Sodeik, B.; Werfel, T. RNase 7 strongly promotes TLR9-mediated DNA sensing by human plasmacytoid dendritic cells. J. Investig. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kopfnagel, V.; Wittmann, M.; Werfel, T. Human keratinocytes express AIM2 and respond to dsDNA with IL-1β secretion. Exp. Dermatol. 2011, 20, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Göblös, A.; Danis, J.; Vas, K.; Bata-Csörgő, Z.; Kemény, L.; Széll, M. Keratinocytes express functional CARD18, a negative regulator of inflammasome activation, and its altered expression in psoriasis may contribute to disease pathogenesis. Mol. Immunol. 2016, 73, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sayama, K.; Tohyama, M.; Shirakata, Y.; Yang, L.; Hirakawa, S.; Tokumaru, S.; Hashimoto, K. The NF-κB, p38 MAPK and STAT1 pathways differentially regulate the dsRNA-mediated innate immune responses of epidermal keratinocytes. Int. Immunol. 2008, 20, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Luff, J.A.; Yuan, H.; Suter, M.M.; Müller, E.J.; Schlegel, R.; Moore, P.F. Canine keratinocytes upregulate type I interferons and proinflammatory cytokines in response to poly(dA:dT) but not to canine papillomavirus. Vet. Immunol. Immunopathol. 2013, 153, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Danis, J.; Göblös, A.; Bata-Csörgő, Z.; Kemény, L.; Széll, M. PRINS non-coding RNA regulates nucleic acid-induced innate immune responses of human keratinocytes. Front. Immunol. 2017, 8, 1053. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F. V Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I dependent sensing of poly(dA-dT) via the induction of an RNA polymerase III transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, D.; Ning, W.; Xu, A. Cytosolic dsDNA triggers apoptosis and pro-inflammatory cytokine production in normal human melanocytes. Exp. Dermatol. 2015, 24, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.-W.; Jiang, R.-H.; Kim, K.-D.; Lee, J.-H.; Kim, C.-D.; Yin, W.-T.; Lee, J.-H. Rosmarinic acid inhibits poly(I:C)-induced inflammatory reaction of epidermal keratinocytes. Life Sci. 2016, 155, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-M.; Choi, D.-K.; Sohn, K.-C.; Kim, S.-Y.; Min Ha, J.; Ho Lee, Y.; Im, M.; Seo, Y.-J.; Deok Kim, C.; Lee, J.-H.; et al. Double-stranded RNA induces inflammation via the NF-κB pathway and inflammasome activation in the outer root sheath cells of hair follicles. Sci. Rep. 2017, 7, 44127. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Tax, G.; Urbán, E.; Palotás, Z.; Puskás, R.; Kónya, Z.; Bíró, T.; Kemény, L.; Szabó, K. Propionic acid produced by Propionibacterium acnes strains contributes to their pathogenicity. Acta Derm. Venereol. 2016, 96, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.A.; Hengeltraub, S.F.; Gao, F.C.; Leivant, M.A.; Spandau, D.F. Aberrant NF-κB activity in HaCaT cells alters their response to UVB signaling. J. Investig. Dermatol. 2006, 126, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Barbuto, S.; Idoyaga, J.; Villa-Perelló, M.; Longhi, M.P.; Breton, G.; Steinman, R.M.; Miur, T.W. Induction of innate and adaptive immunity by delivery of poly dA:dT to dendritic cells. Nat. Chem. Biol. 2013, 9, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kis-Toth, K.; Szanto, A.; Thai, T.-H.; Tsokos, G.C. Cytosolic DNA-activated human dendritic cells are potent activators of the adaptive immune response. J. Immunol. 2011, 187, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.-H.; Homey, B.; Cao, W.; Wang, Y.-H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Oláh, A.; Ambrus, L.; Nicolussi, S.; Gertsch, J.; Tubak, V.; Kemény, L.; Soeberdt, M.; Abels, C.; Bíró, T. Inhibition of fatty acid amide hydrolase exerts cutaneous anti-inflammatory effects both in vitro and in vivo. Exp. Dermatol. 2016, 25, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Szegedi, K.; Göblös, A.; Bacsa, S.; Antal, M.; Németh, I.B.; Bata-Csörgő, Z.; Kemény, L.; Dobozy, A.; Széll, M. Expression and functional studies on the noncoding RNA, PRINS. Int. J. Mol. Sci. 2013, 14, 205–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Sayama, K.; Yamasaki, K.; Tohyama, M.; Shirakata, Y.; Hanakawa, Y.; Tokumaru, S.; Yahata, Y.; Yang, L.; Yoshimura, A.; et al. SOCS1-negative feedback of STAT1 activation is a key pathway in the dsRNA-induced innate immune response of human keratinocytes. J. Investig. Dermatol. 2006, 126, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Hemmi, H.; Akira, S. Interferon response induced by Toll-like receptor signaling. J. Endotoxin Res. 2004, 10, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Scherle, P.A.; Jones, E.A.; Favata, M.F.; Daulerio, A.J.; Covington, M.B.; Nurnberg, S.A.; Magolda, R.L.; Trzaskos, J.M. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J. Immunol. 1998, 161, 5681–5686. [Google Scholar] [PubMed]
- Jaffee, B.D.; Manos, E.J.; Collins, R.J.; Czerniak, P.M.; Favata, M.F.; Magolda, R.L.; Scherle, P.A.; Trzaskos, J.M. Inhibition of MAP kinase kinase (MEK) results in an anti-inflammatory response in vivo. Biochem. Biophys. Res. Commun. 2000, 268, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Chan, J.H.P.; Wong, C.H.; Leung, B.P.; Wong, W.S.F. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J. Immunol. 2004, 172, 7053–7059. [Google Scholar] [CrossRef] [PubMed]
- Lulli, D.; Carbone, M.; Pastore, S. The MEK inhibitors trametinib and cobimetinib induce a type I interferon response in human keratinocytes. Int. J. Mol. Sci. 2017, 18, 2227. [Google Scholar] [CrossRef] [PubMed]
- Maeng, Y.S.; Min, J.K.; Kim, J.H.; Yamagishi, A.; Mochizuki, N.; Kwon, J.Y.; Park, Y.W.; Kim, Y.M.; Kwon, Y.G. ERK is an anti-inflammatory signal that suppresses expression of NF-κB-dependent inflammatory genes by inhibiting IKK activity in endothelial cells. Cell. Signal. 2006, 18, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Lulli, D.; Carbone, M.L.; Pastore, S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget 2016, 7, 47777–47793. [Google Scholar] [CrossRef] [PubMed]
- Luecke, S.; Paludan, S.R. Molecular requirements for sensing of intracellular microbial nucleic acids by the innate immune system. Cytokine 2016, 98, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Andrés, R.M.; Hald, A.; Johansen, C.; Kragballe, K.; Iversen, L. Studies of Jak/STAT3 expression and signalling in psoriasis identifies STAT3-Ser727 phosphorylation as a modulator of transcriptional activity. Exp. Dermatol. 2013, 22, 323–328. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danis, J.; Janovák, L.; Gubán, B.; Göblös, A.; Szabó, K.; Kemény, L.; Bata-Csörgő, Z.; Széll, M. Differential Inflammatory-Response Kinetics of Human Keratinocytes upon Cytosolic RNA- and DNA-Fragment Induction. Int. J. Mol. Sci. 2018, 19, 774. https://doi.org/10.3390/ijms19030774
Danis J, Janovák L, Gubán B, Göblös A, Szabó K, Kemény L, Bata-Csörgő Z, Széll M. Differential Inflammatory-Response Kinetics of Human Keratinocytes upon Cytosolic RNA- and DNA-Fragment Induction. International Journal of Molecular Sciences. 2018; 19(3):774. https://doi.org/10.3390/ijms19030774
Chicago/Turabian StyleDanis, Judit, Luca Janovák, Barbara Gubán, Anikó Göblös, Kornélia Szabó, Lajos Kemény, Zsuzsanna Bata-Csörgő, and Márta Széll. 2018. "Differential Inflammatory-Response Kinetics of Human Keratinocytes upon Cytosolic RNA- and DNA-Fragment Induction" International Journal of Molecular Sciences 19, no. 3: 774. https://doi.org/10.3390/ijms19030774
APA StyleDanis, J., Janovák, L., Gubán, B., Göblös, A., Szabó, K., Kemény, L., Bata-Csörgő, Z., & Széll, M. (2018). Differential Inflammatory-Response Kinetics of Human Keratinocytes upon Cytosolic RNA- and DNA-Fragment Induction. International Journal of Molecular Sciences, 19(3), 774. https://doi.org/10.3390/ijms19030774