The Inside Story of Adenosine


Abstract
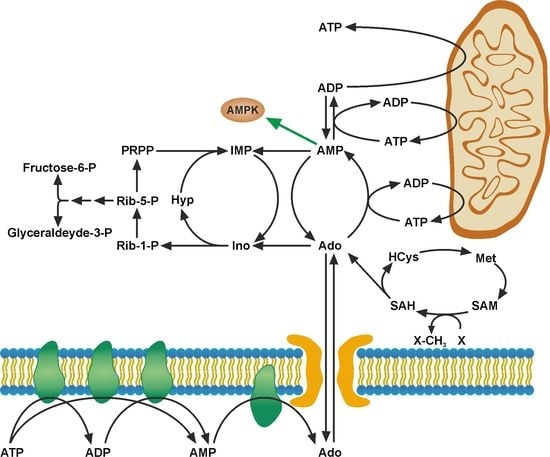
:
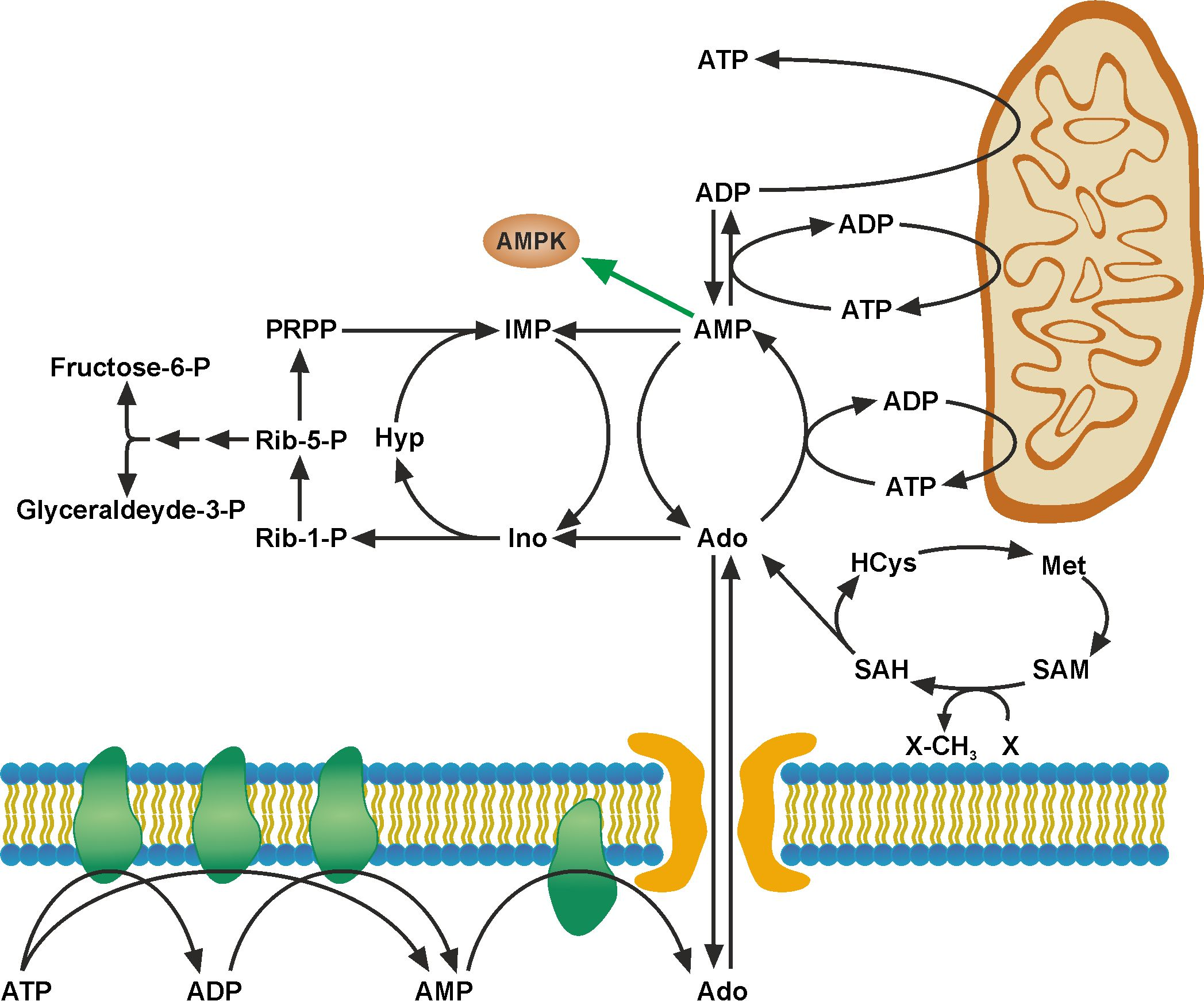{kind=link}
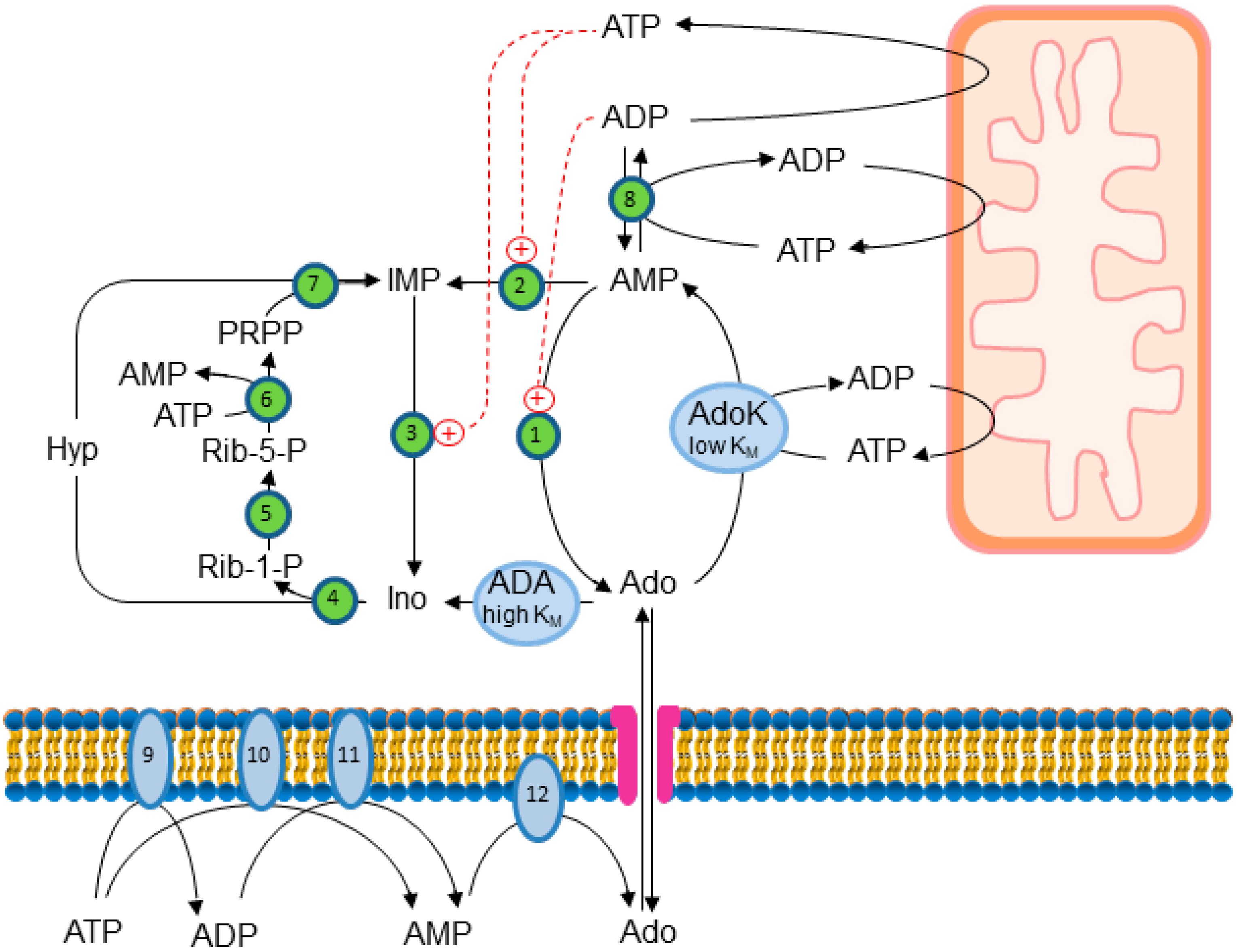{kind=link}
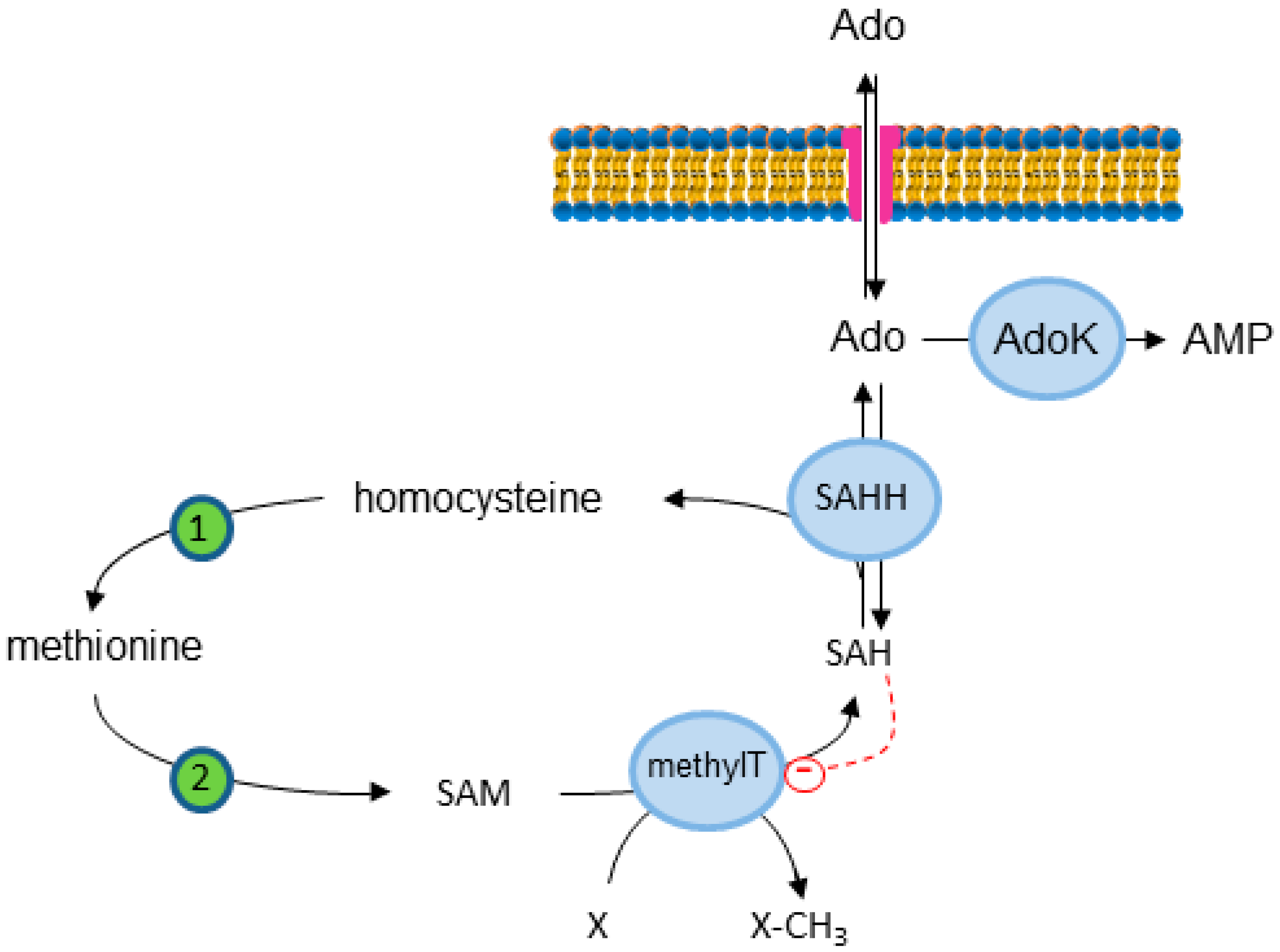{kind=link}
{kind=link}
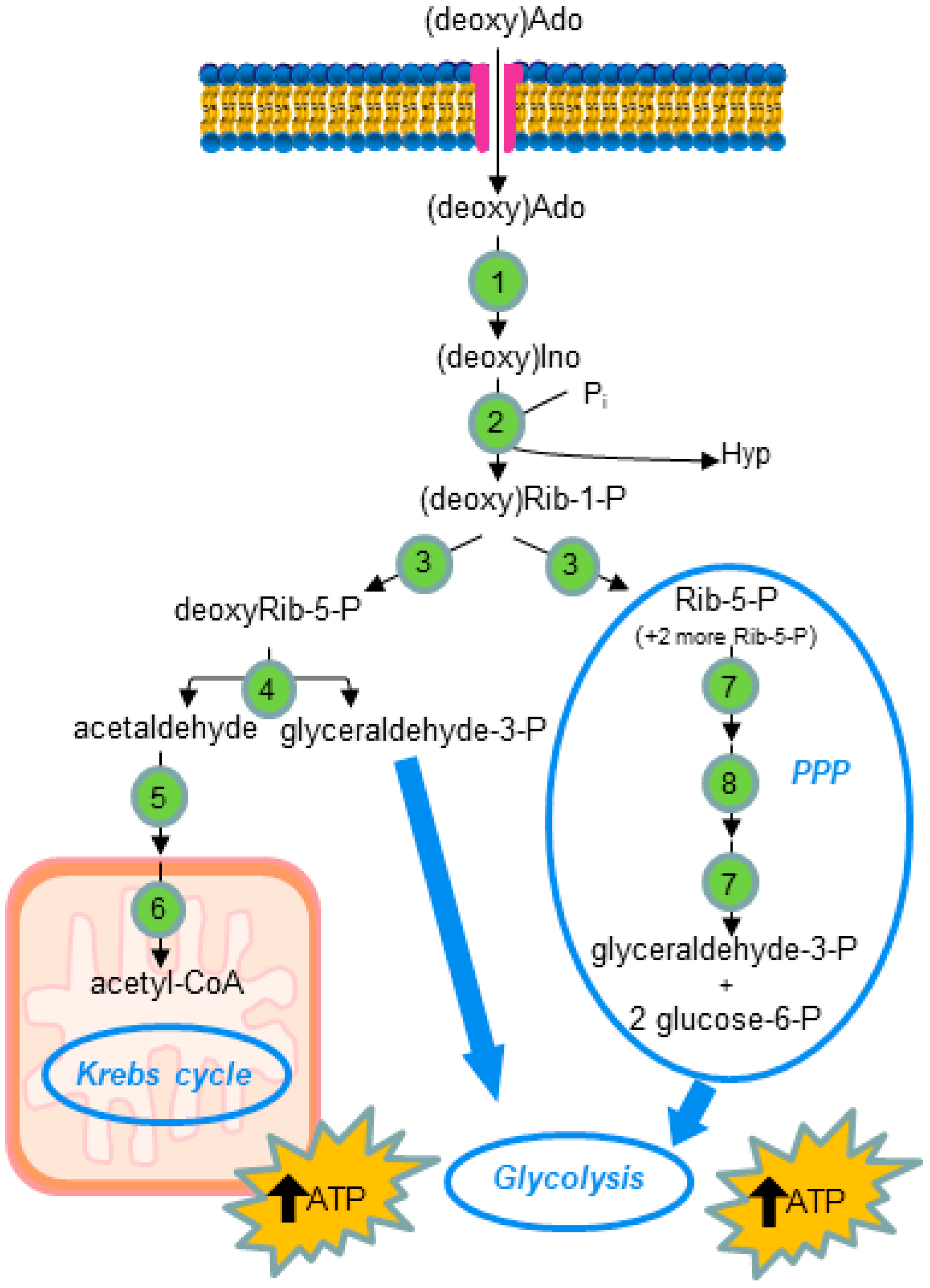
1. Introduction
2. Hypothermia Induced by Adenylate Compounds
3. Intracellular Ado in the Central Nervous System
3.1. Transmethylation Pathway
3.2. Sleep Homeostasis
4. Regulatory Mechanisms Mediated by dAdo
5. Ado as an Energy Source
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ipata, P.L.; Balestri, F.; Camici, M.; Tozzi, M.G. Molecular mechanisms of nucleoside recycling in the brain. Int. J. Biochem. Cell Biol. 2011, 43, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ipata, P.L.; Pesi, R. Nucleoside recycling in the brain and the nucleosidome: A complex metabolic and molecular cross-talk between the extracellular nucleotide cascade system and the intracellular nucleoside salvage. Metabolomics 2016, 12, 22. [Google Scholar] [CrossRef]
- Ashby, B.; Holmsen, H. Platelet AMP deaminase: Regulation by Mg–ATP2- and inorganic phosphate and inhibition by the transition state analog coformycin. J. Biol. Chem. 1983, 258, 3668–3672. [Google Scholar] [PubMed]
- Tozzi, M.G.; Pesi, R.; Allegrini, S. On the physiological role of cytosolic 5′-nucleotidase II (cN-II): Pathological and therapeutical implications. Curr. Med. Chem. 2013, 20, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell. Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skladanowski, A.C.; Newby, A.C. Partial purification and properties of an AMP-specific soluble 5′-nucleotidase from pigeon heart. Biochem. J. 1990, 268, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ford, H., Jr.; Dai, F.; Mu, L.; Siddiqui, M.A.; Nicklaus, M.C.; Anderson, L.; Marquez, V.E.; Barchi, J.J., Jr. Adenosine deaminase prefers a distinct sugar ring conformation for binding and catalysis: Kinetic and structural studies. Biochemistry 2000, 39, 2581–2592. [Google Scholar] [CrossRef] [PubMed]
- Ipata, P.L.; Camici, M.; Micheli, V.; Tozzi, M.G. Metabolic network of nucleosides in the brain. Curr. Top. Med. Chem. 2011, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.A.; Beal, P.R.; Yao, S.Y.; King, A.E.; Cass, C.E.; Young, J.D. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004, 447, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, I.; Pastor-Anglada, M.; Casado, F.J. Long term endocrine regulation of nucleoside transporters in rat intestinal epithelial cells. J. Gen. Physiol. 2004, 124, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Arslan, G.; Halldner, L.; Kull, B.; Schulte, G.; Wasserman, W. Structure and function of adenosine receptors and their genes. Naunyn-Schmiedebergs Arch. Pharmacol. 2000, 362, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Van Calker, D.; Muller, M.; Hamprecht, B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J. Neurochem. 1979, 33, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W.; Butts-Lamb, P.; Padgett, W. Subclasses of adenosine receptors in the central nervous system: Interaction with caffeine and related methylxanthines. Cell. Mol. Neurobiol. 1983, 3, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signaling in the cardiovascular system. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Kulkarni, S.K. Effect of purinergic substances on rectal temperature in mice: Involvement of P1-purinoceptors. Arch. Int. Pharmacodyn. Ther. 1983, 264, 180–186. [Google Scholar] [PubMed]
- Olson, J.M.; Jinka, T.R.; Larson, L.K.; Danielson, J.J.; Moore, J.T.; Carpluck, J.; Drew, K.L. Circannual rhythm in body temperature, torpor, and sensitivity to A1 adenosine receptor agonist in arctic ground squirrels. J. Biol. Rhythms 2013, 28, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Carlin, J.L.; Jain, S.; Gizewski, E.; Wan, T.C.; Tosh, D.K.; Xiao, C.; Auchampach, J.A.; Jacobson, K.A.; Gavrilova, O.; Reitman, M.L. Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacology 2017, 114, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Rittiner, J.E.; Korboukh, I.; Hull-Ryde, E.A.; Jin, J.; Janzen, W.P.; Frye, S.V.; Zylka, M.J. AMP is an adenosine A1 receptor agonist. J. Biol. Chem. 2012, 287, 5301–5309. [Google Scholar] [CrossRef] [PubMed]
- Muzzi, M.; Blasi, F.; Masi, A.; Coppi, E.; Traini, C.; Felici, R.; Piattelli, M.; Cavone, L.; Pugliese, A.M.; Moroni, F.; et al. Neurological basis of AMP-dependent thermoregulation and its relevance to central and peripheral hypothermia. J. Cereb. Blood Flow Metab. 2009, 33, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kaasik, K.; Blackburn, M.R.; Lee, C.C. Constant darkness is a circadian metabolic signal in mammals. Nature 2006, 439, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Eisner, C.; Kim, S.; Grill, A.; Qin, Y.; Hoerl, M.; Briggs, J.; Castrop, H.; Thiel, M.; Schnermann, J. Profound hypothermia after adenosine kinase inhibition in A1AR deficient mice suggests a receptor-independent effect of intracellular adenosine. Pflugers Arch. 2017, 469, 339–3347. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets-what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem. Int. 2001, 38, 107–125. [Google Scholar] [CrossRef]
- Boison, D. Adenosine kinase: Exploitation for therapeutic gain. Pharmacol. Rev. 2013, 65, 906–943. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B. Rethinking the purinergic neuron-glia connection. Proc. Natl. Acad. Sci. USA 2012, 109, 5913–5914. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Maysami, S.; Studer, F.E.; Wilz, A.; Simon, R.P.; Boison, D. Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J. Cereb. Blood Flow Metab. 2008, 28, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Adenosinergic signaling in epilepsy. Neuropharmacology 2016, 104, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ren, G.; Lusardi, T.; Wilz, A.; Lan, J.Q.; Iwasato, T.; Itohara, S.; Simon, R.P.; Boison, D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J. Clin. Investig. 2008, 118, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [PubMed]
- Drabikowska, A.K.; Halec, L.; Shugar, D. Purification and properties of adenosine kinase from rat liver: Separation from deoxyadenosine kinase activity. Z. Naturforsch C 1985, 40, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; McGarrity, S.; Shen, H.Y.; Boison, D.; Yee, B.K. Working memory and the homeostatic control of brain adenosine by adenosine kinase. Neuroscience 2012, 213, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Sandau, U.S.; Colino-Oliveira, M.; Jones, A.; Saleumvong, B.; Coffman, S.Q.; Liu, L.; Miranda-Lourenço, C.; Palminha, C.; Batalha, V.L.; Xu, Y.; et al. Adenosine kinase deficiency in the brain results in maladaptive synaptic plasticity. J. Neurosci. 2016, 36, 12117–12128. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Adenosine as a modulator of brain activity. Drug News Perspect. 2007, 20, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investig. 2013, 123, 3552–3563. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-Y.; Lusardi, T.A.; Williams-Karnesky, R.L.; Lan, J.Q.; Poulsen, D.J.; Boison, D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J. Cereb. Blood Flow Metab. 2011, 31, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Adair, T.H. Growth regulation of the vascular system: An emerging role for adenosine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283–R296. [Google Scholar] [CrossRef] [PubMed]
- Feoktistov, I.; Biaggioni, I.; Cronstein, B.N. Adenosine receptors in wound healing, fibrosis and angiogenesis. Handb. Exp. Pharmacol. 2009, 193, 383–397. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Y.; Yan, S.; Zhou, Y.; Yang, Q.; Pan, Y.; Zeng, X.; An, X.; Liu, Z.; Wang, L.; et al. Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol. Med. 2017, 9, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Decking, U.K.; Schlieper, G.; Kroll, K.; Schrader, J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 1997, 81, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Morote-Garcia, J.C.; Rosenberger, P.; Kuhlicke, J.; Eltzschig, H.K. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 2008, 111, 5571–5580. [Google Scholar] [CrossRef] [PubMed]
- Studer, F.E.; Fedele, D.E.; Marowsky, A.; Schwerdel, C.; Wernli, K.; Vogt, K.; Fritschy, J.M.; Boison, D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 2006, 142, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Florian, C.; Fellin, T.; Munoz, J.R.; Lee, S.Y.; Abel, T.; Haydon, P.G.; Frank, M.G. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 2009, 61, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, M.R.; Taishi, P.; Clinton, J.M.; Krueger, J.M. 5-Ectonucleotidase- knockout mice lack non-REM sleep responses to sleep deprivation. Eur. J. Neurosci. 2012, 35, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Palchykova, S.; Winsky-Sommerer, R.; Shen, H.Y.; Boison, D.; Gerling, A.; Tobler, I. Manipulation of adenosine kinase affects sleep regulation in mice. J. Neurosci. 2010, 30, 13157–13165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radek, R.J.; Decker, M.W.; Jarvis, M.F. The adenosine kinase inhibitor ABT-702 augments EEG slow waves in rats. Brain Res. 2004, 1026, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Bjorness, T.E.; Dale, N.; Mettlach, G.; Sonneborn, A.; Sahin, B.; Fienberg, A.A.; Yanagisawa, M.; Bibb, J.A.; Greene, R.W. An adenosine-mediated glial-neuronal circuit for homeostatic sleep. J. Neurosci. 2016, 36, 3709–3721. [Google Scholar] [CrossRef] [PubMed]
- Greene, R.W.; Bjorness, T.E.; Suzuki, A. The adenosine-mediated, neuronal-glial, homeostatic sleep response. Curr. Opin. Neurobiol. 2017, 44, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Carson, D.A.; Kaye, J.; Seegmiller, J.E. Lymphospecific toxicity in adenosine-deaminase deficiency and purine nucleoside-phosphorylase deficiency: Posible role of nucleoside kinase(s). Proc. Natl. Acad. Sci. USA 1977, 74, 5677–5681. [Google Scholar] [CrossRef] [PubMed]
- Takeda, E.; Kuroda, Y.; Naito, E.; Yokota, I.; Saijo, T.; Hirose, M.; Miyao, M. Effects of deoxyadenosine on ribonucleotide reductase in adenosine-deaminase deficient lymphocytes. J. Inherit. Metabol. Dis. 1991, 14, 87–95. [Google Scholar] [CrossRef]
- Agarwal, S.P.; Spector, T.; Parks, R.E., Jr. Tight-binding inhibitors. IV. Inhibition of adenosine deaminase by various inhibitors. Biochem. Pharmacol. 1977, 26, 359–367. [Google Scholar] [CrossRef]
- Döhner, H.; Ho, A.D.; Thaler, J.; Stryckmans, P.; Sonneveld, P.; de Witte, T.; Lechner, K.; Lauria, F.; Bödewadt-Radzun, S.; Suciu, S.; et al. Pentostatin in polymphocytic leukemia: Phase-II trial of the European Organization for research and treatment of cancer leukemia cooperative study group. J. Nat. Cancer Inst. 1993, 85, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Rossi, D.; Motta, M.; Quaresmini, G.; Rossi, M.; Coscia, M.; Anastasia, A.; Rossini, F.; Cortelezzi, A.; Nador, G.; et al. A phase II multi-center trial of pentostatin plus cyclophosphamide with ofatumumab in older previously untreated chronic lymphocytic leukemia patients. Haematologica 2015, 100, E501–E504. [Google Scholar] [CrossRef] [PubMed]
- Lapi, L.; Cohen, S.S. Toxicities of adenosine and 2′-deoxyadenosine in L cells treated with inhibitors of adenosine deaminase. Biochem. Pharmacol. 1977, 26, 71–76. [Google Scholar] [CrossRef]
- Hunt, S.W., III; Hoffee, P.A. Adenosine deaminase from deoxycoformycin-sensitive and -resistant rat hepatoma cells. Purification and characterization. J. Biol. Chem. 1982, 257, 14239–14244. [Google Scholar] [PubMed]
- Camici, M.; Turriani, M.; Tozzi, M.G.; Turchi, G.; Cos, J.; Alemany, C.; Miralles, A.; Noe, V.; Ciudad, C.J. Purine enzyme profile in human colon-carcinoma cell lines and differential sensitivity to deoxycoformycin and 2′deoxyadenosine in combination. Int. J. Cancer 1995, 62, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Bemi, V.; Tazzini, N.; Banditelli, S.; Giorgelli, F.; Pesi, R.; Turchi, G.; Mattana, A.; Sgarrella, F.; Tozzi, M.G.; Camici, M. Deoxyadenosine metabolism in a human colon-carcinoma cell line (LoVo) in relation to its cytotoxic effect in combination with deoxycoformycin. Int. J. Cancer 1998, 75, 713–720. [Google Scholar] [CrossRef]
- Giannecchini, M.; D’Innocenzo, B.; Pesi, R.; Sgarrella, F.; Iorio, M.; Collecchi, P.; Tozzi, M.G.; Camici, M. 2′-deoxyadenosine causes apoptotic cell death in a human colon carcinoma cell line. J. Biochem. Mol. Toxicol. 2003, 17, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.S.; Wakade, A.R. Quantitative analysis of similarities and differences caused by adenosine and 2′-deoxyadenosine in sympathetic neurons. J. Neurochem. 1996, 67, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Wakade, A.R.; Guo, X.; Palmer, K.C.; Kulkarni, J.S.; Przywara, D.A.; Wakade, T.D. 2′-deoxyadenosine induces apoptosis in rat chromaffin cells. J. Neurochem. 1996, 67, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Unsiker, K. The chromaffin cell: Paradigm in cell, developmental and growth factor biology. J. Anat. 1993, 183, 207–221. [Google Scholar]
- Garcia-Gil, M.; Tozzi, M.G.; Allegrini, S.; Folcarelli, S.; Della Sala, G.; Voccoli, V.; Colombaioni, L.; Camici, M. Novel metabolic aspects related to adenosine deaminase inhibition in a human astrocytoma cell line. Neurochem. Int. 2012, 60, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Tozzi, M.G.; Varani, S.; Della Verde, L.; Petrotto, E.; Balestri, F.; Colombaioni, L.; Camici, M. The combination of adenosine deaminase inhibition and deoxyadenosine induces apoptosis in a human astrocytoma cell line. Neurochem. Int. 2015, 80, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Tozzi, M.G.; Balestri, F.; Colombaioni, L.; Camici, M. Mitochondrial damage and apoptosis induced by adenosine deaminase inhibition and deoxyadenosine in human neuroblastoma cell lines. J. Cell. Biochem. 2016, 117, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Micheli, V.; Ipata, P.L.; Tozzi, M.G. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem. Int. 2010, 56, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Sauer, A.V.; Hernandez, R.J.; Fumagalli, F.; Bianchi, V.; Poliani, P.L.; Dallatomasina, C.; Riboni, E.; Politi, L.S.; Tabucchi, A.; Carlucci, F.; et al. Alterations in the brain adenosine metabolism cause neurological impairment in ADA-deficient mice and patients. Sci. Rep. 2017, 7, 40136. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Does impairment of energy metabolism result in exocitotoxic neuronal death in neurodegenerative illness? Ann. Neurol. 1992, 50, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, F.E.; Xiong, W. Stimulus- and cell-type-specific release of purines in cultured rat forebrain astrocytes and neurons. J. Neurochem. 2004, 88, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Rudolphi, K.A.; Shubert, P. Adenosine and brain ischemia. In Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology; Belardinelli, L., Pelleg, A., Eds.; Springer: Boston, MA, USA, 1995; pp. 391–397. ISBN 978-1-4615-2011-5. [Google Scholar]
- Ciccarelli, R.; di Iorio, P.; Giuliani, P.; D’Alimonte, I.; Ballerini, P.; Caciagli, F.; Rathbone, M.P. Rat cultured astrocytes release guanine-based purines in basal conditions and after hypoxia/hypoglycemia. Glia 1999, 25, 93–98. [Google Scholar] [CrossRef]
- Böcklinger, K.; Tomaselli, B.; Heftberger, V.; Podhraski, V.; Bandtlow, C.; Baier-Bitterlich, G. Purine nucleosides support the neurite outgrowth of primary rat cerebellar granule cells after hypoxia. Eur. J. Cell Biol. 2004, 83, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Haun, S.E.; Segeleon, J.E.; Trapp, V.L.; Clotz, M.A.; Horrocks, L.A. Inosine mediates the protective effect of adenosine in rat astrocyte cultures subjected to combined glucose-oxygen deprivation. J. Neurochem. 1996, 67, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Jurkowitz, M.S.; Litsky, M.J.; Browning, M.J.; Hohl, C.M. Adenosine, inosine and guanosine protect glial cells during glucose deprivation and mitochondrial inhibition: Correlation between protection and ATP preservation. J. Neurochem. 1998, 71, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Litsky, M.L.; Hohl, C.M.; Lucas, J.H.; Jurkowitz, M.S. Inosine and guanosine preserve neuronal and glial cell viability in mouse spinal cord cultures during chemical hypoxia. Brain Res. 1999, 821, 426–432. [Google Scholar] [CrossRef]
- Yoo, B.-K.; Choi, J.W.; Yoon, S.Y.; Ko, K.H. Protective effect of adenosine and purine nucleos(t)ides against the death by hydrogen peroxide and glucose deprivation in rat primary astrocytes. Neurosci. Res. 2005, 51, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Giannecchini, M.; Sgarrella, F.; Carta, M.C.; Tozzi, M.G.; Camici, M. Purine and pyrimidine nucleosides preserve human astrocytoma cell adenylate energy charge under ischemic conditions. Neurochem. Int. 2007, 50, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Giannecchini, M.; Matteucci, M.; Pesi, R.; Sgarrella, F.; Tozzi, M.G.; Camici, M. Uptake and utilization of nucleosides for energy repletion. Int. J. Biochem. Cell Biol. 2005, 37, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Wikström, G.; Kavianipour, M.; Ronquist, G.; Waldenström, A. Pre-conditioning activates adenosine utilization in a cost-effective way during myocardial ischaemia. Acta Physiol. Scand. 2001, 173, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Gerő, D.; Stangl, R.; Rosero, O.; Szijártó, A.; Lotz, G.; Mohácsik, P.; Szoleczky, P.; Coletta, C.; Szabó, C. Adenosine and inosine exert cytoprotective effects in an in vitro model of liver ischemia-reperfusion injury. Int. J. Mol. Med. 2013, 31, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bzowska, A.; Kulikowska, E.; Shugar, D. Purine nucleoside phosphorylases: Properties, functions, and clinical aspects. Pharmacol. Ther. 2000, 88, 349–425. [Google Scholar] [CrossRef]
- Sgarrella, F.; Poddie, F.P.A.; Meloni, M.A.; Sciola, L.; Pipia, P.; Tozzi, M.G. Channeling of deoxyribose moiety of exogenous DNA into carbohydrate metabolism: Role of deoxyriboaldolase. Comp. Biochem. Physiol. 1997, 117B, 253–257. [Google Scholar] [CrossRef]
- Moriwaki, Y.; Yamamoto, T.; Yamakita, J.; Takahashi, S.; Higashino, K. Comparative localization of aldehyde oxidase and xanthine oxidoreductase activity in rat tissues. Histochem. J. 1998, 30, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.; Gracia, E.; Moreno, E.; Mallol, J.; Lluís, C.; Canela, E.I.; Casadó, V. Moonlighting adenosine deaminase: A target protein for drug development. Med. Res. Rev. 2015, 35, 85–125. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camici, M.; Garcia-Gil, M.; Tozzi, M.G. The Inside Story of Adenosine. Int. J. Mol. Sci. 2018, 19, 784. https://doi.org/10.3390/ijms19030784
Camici M, Garcia-Gil M, Tozzi MG. The Inside Story of Adenosine. International Journal of Molecular Sciences. 2018; 19(3):784. https://doi.org/10.3390/ijms19030784
Chicago/Turabian StyleCamici, Marcella, Mercedes Garcia-Gil, and Maria Grazia Tozzi. 2018. "The Inside Story of Adenosine" International Journal of Molecular Sciences 19, no. 3: 784. https://doi.org/10.3390/ijms19030784
APA StyleCamici, M., Garcia-Gil, M., & Tozzi, M. G. (2018). The Inside Story of Adenosine. International Journal of Molecular Sciences, 19(3), 784. https://doi.org/10.3390/ijms19030784