Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels

 ,
,  ,
,
Abstract
:1. Introduction
2. Coronary Blood Flow Regulation
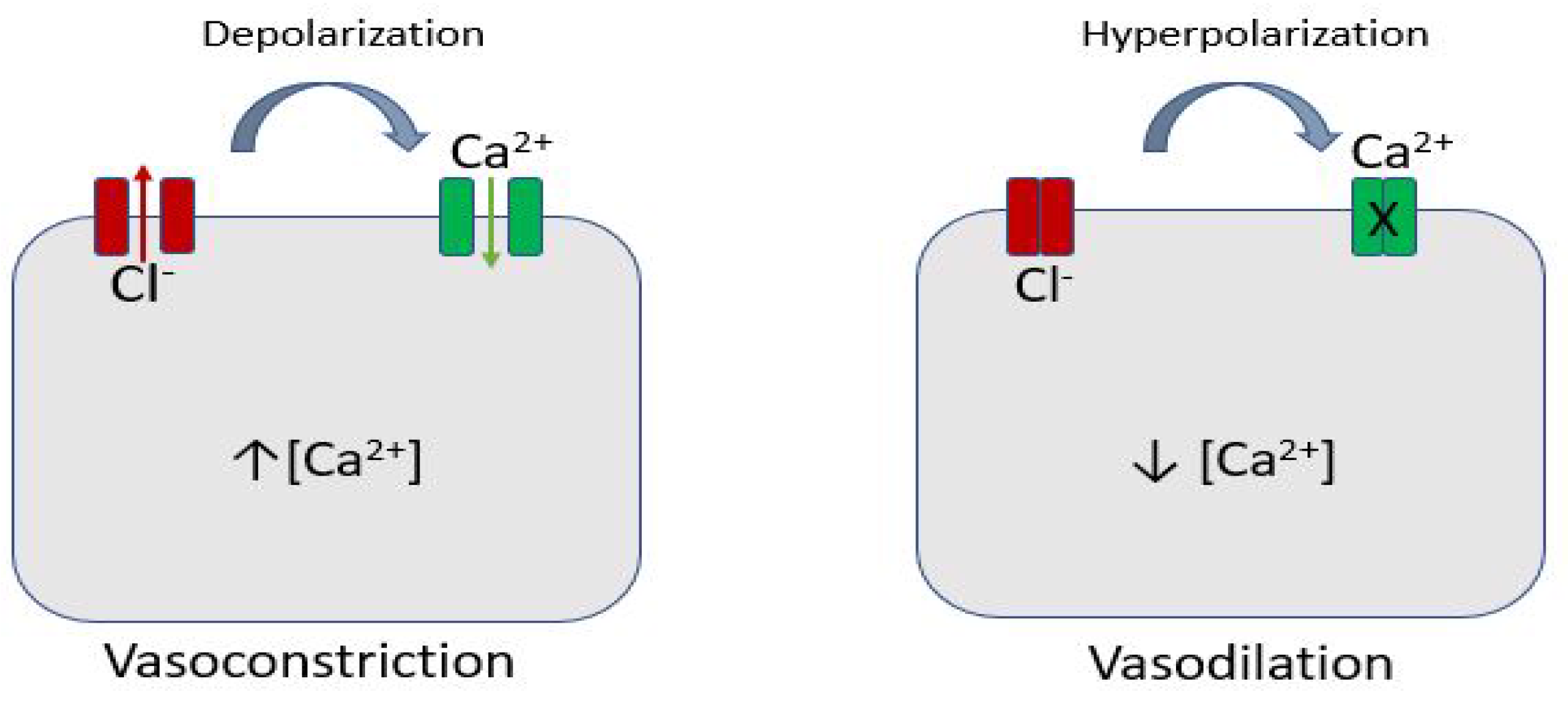
3. Role of Ion Channels in Coronary Blood Flow Regulation
4. Cardiovascular Risk Factors and Ischemic Heart Disease
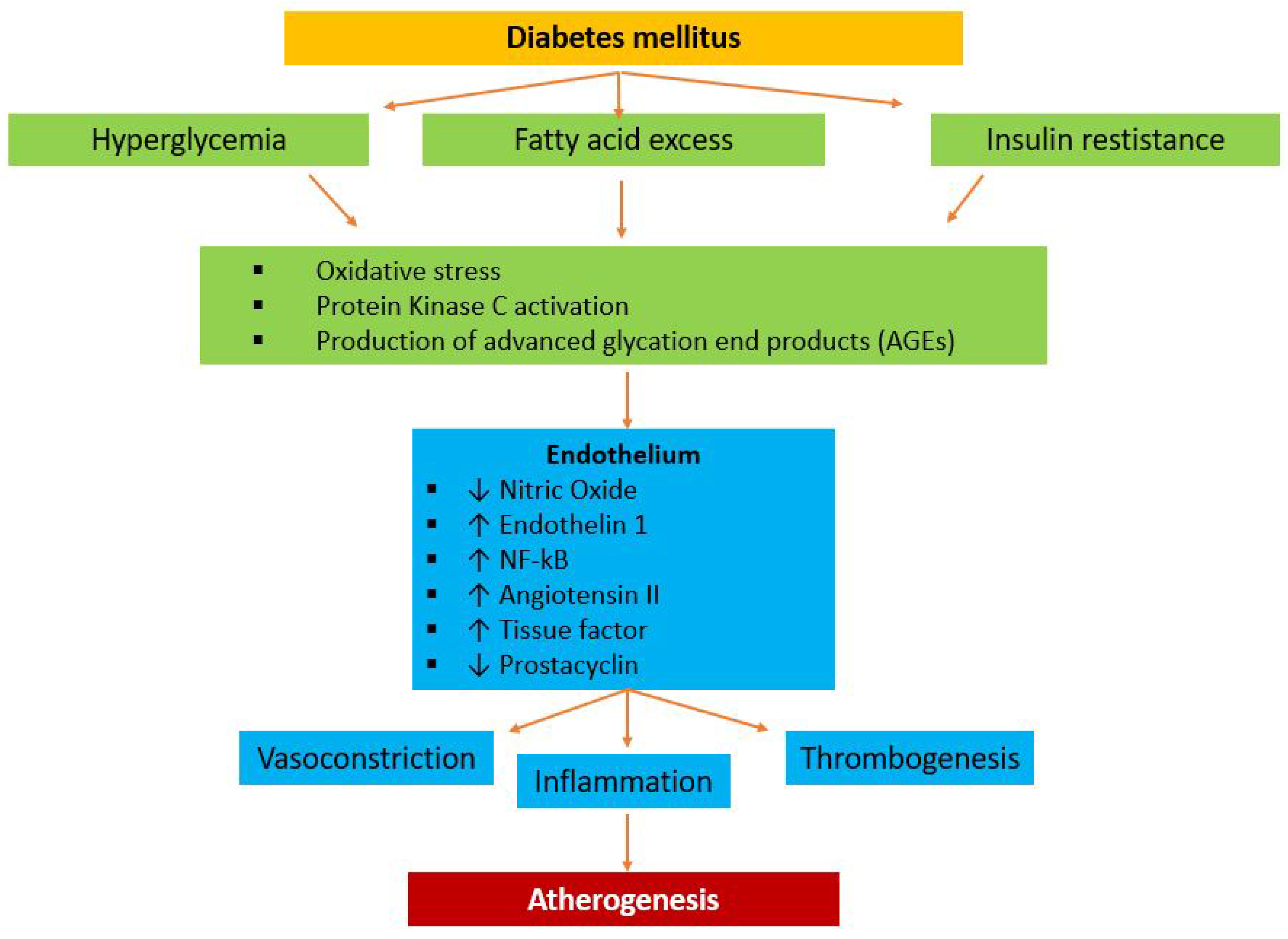
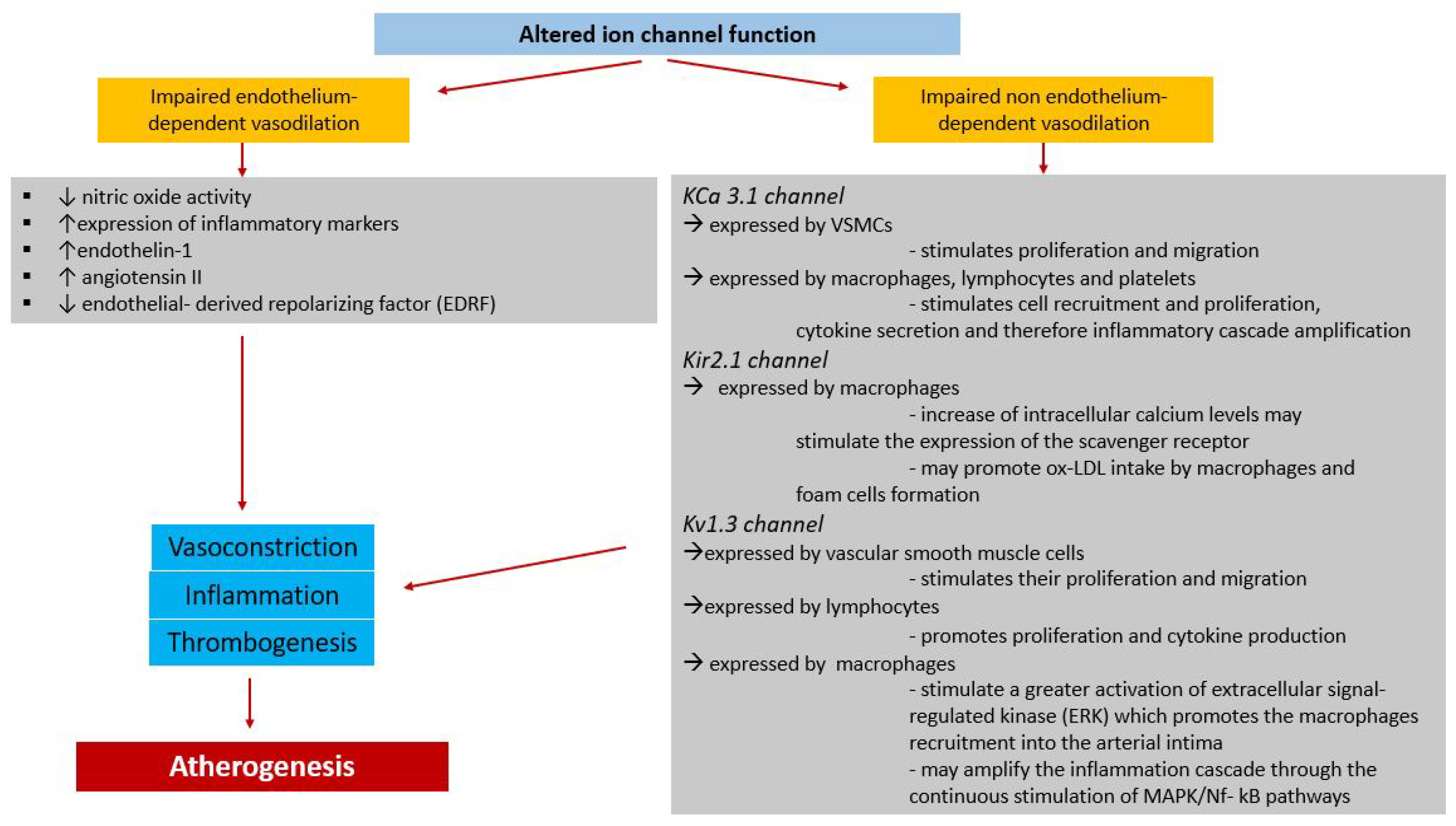
5. Role of Ion Channels in the Pathophysiological Continuum between Cardiovascular Risk Factors and Ischemic Heart Disease
6. Conclusions
Conflicts of Interest
Abbreviations
| DM | diabetes mellitus |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
| MVO2 | myocardial oxygen consumption |
| NO | nitric oxide |
| K-ATP | triphosphate adenosine-sensitive potassium channels |
| KCa | calcium-dependent potassium channels |
| Kv | voltage dependent potassium channels |
| Kir | inward-rectifier potassium channel |
| Nav | voltage-gated sodium channels |
| Cav | voltage-gated calcium channels |
| VSMCs | vascular smooth muscle cells |
| AGEs | advanced glycation products |
| ECs | endothelial cells |
| ox-LDL | oxidized LDL |
| ROSIHD | reactive oxygen speciesischemic heart disease |
| CAD | coronary heart disease |
| SNPs | single-nucleotide polymorphisms |
References
- World Health Organization. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Fedele, F.; Severino, P.; Bruno, N.; Stio, R.; Caira, C.; D’Ambrosi, A.; Brasolin, B.; Ohanyan, V.; Mancone, M. Role of ion channels in coronary microcirculation: A review of the literature. Future Cardiol. 2013, 9, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Goodwill, A.G.; Dick, G.M.; Kiel, A.M. Regulation of Coronary Blood Flow. Compr. Physiol. 2017, 7, 321–382. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Olivotto, I.; Rimoldi, O.E. The coronary circulation and blood flow in left ventricular hypertrophy. J. Mol. Cell. Cardiol. 2012, 52, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E. Adolf Fick–forgotten genius of cardiology. Am. J. Cardiol. 1972, 30, 662–665. [Google Scholar] [CrossRef]
- Berwick, Z.C.; Moberly, S.P.; Kohr, M.C.; Morrical, E.B.; Kurian, M.M.; Dick, G.M.; Tune, J.D. Contribution of voltage-dependent K+ and Ca2+ channels to coronary pressure-flow autoregulation. Basic Res. Cardiol. 2012, 107, 264. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Battaglia, A.; Grossini, E.; Mary, D.A.S.G.; Bona, G.; Scott, E.; Vacca, G. Effects of insulin on coronary blood flow in anesthetized pigs. J. Vasc. Res. 2002, 39, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Lamping, K.G.; Kanatsuka, H.; Eastham, C.L.; Chilian, W.M.; Marcus, M.L. Nonuniform vasomotor responses of the coronary microcirculation to serotonin and vasopressin. Circ. Res. 1989, 65, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.J. Histaminergic modulation of coronary vascular resistance: Are we missing a therapeutic adjunct for the treatment of myocardial ischemia? J. Am. Coll. Cardiol. 1991, 17, 346–347. [Google Scholar] [CrossRef]
- Zhang, C.; Knudson, J.D.; Setty, S.; Araiza, A.; Dincer, U.D.; Kuo, L.; Tune, J.D. Coronary arteriolar vasoconstriction to angiotensin II is augmented in prediabetic metabolic syndrome via activation of AT1 receptors. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2154–H2162. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Mancone, M.; Chilian, W.M.; Severino, P.; Canali, E.; Logan, S.; De Marchis, M.L.; Volterrani, M.; Palmirotta, R.; Guadagni, F. Role of genetic polymorphisms of ion channels in the pathophysiology of coronary microvascular dysfunction and ischemic heart disease. Basic Res. Cardiol. 2013, 108, 387. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.M.; Bratz, I.N.; Borbouse, L.; Payne, G.A.; Dincer, U.D.; Knudson, J.D.; Rogers, P.A.; Tune, J.D. Voltage dependent K+ channels regulate the duration of reactive hyperemia in the canine coronary circulation. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2371–H2381. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.; Zhang, C.; Tune, J.D.; Potter, B.; Kiyooka, T.; Rogers, P.A.; Knudson, J.D.; Dick, G.M.; Swafford, A.; Chilian, W.M. Hydrogen peroxide: A feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.B.; Tune, J.D.; Borbouse, L.; Long, X.; Sturek, M.; Laughlin, M.H. Altered mechanism of adenosine-induced coronary arteriolar dilation in early-stage metabolic syndrome. Exp. Biol. Med. 2009, 234, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Berwick, Z.C.; Payne, G.A.; Lynch, B.; Dick, G.M.; Sturek, M.; Tune, J.D. Contribution of adenosine A2A and A2B receptors to ischemic coronary dilation: Role of KV and KATP channels. Microcirculation 2010, 17, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.; Kiyooka, T.; Rocic, P.; Rogers, P.A.; Zhang, C.; Swafford, A.; Dick, G.M.; Viswanathan, C.; Park, Y.; Chilian, W.M. Redox-dependent coronary metabolic dilation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3720–H3725. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, J.; Werner, M.E.; Brayden, J.E.; Nelson, M.T. Calcium-activated potassium channels and the regulation of vascular tone. Physiology 2006, 21, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Kaistha, B.P.; Hoyer, J.; Köhler, R. Endothelial Ca2+-activated K+ channels in normal and impaired EDHF-dilator responses—Relevance to cardiovascular pathologies and drug discovery. Br. J. Pharmacol. 2009, 157, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, Y.H.; Clements, R.T.; Sodha, N.R.; Khabbaz, K.R.; Senthilnathan, V.; Nishimura, K.K.; Alper, S.L.; Sellke, F.W. Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegia arrest. Circulation 2008, 118, S46–S51. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.M.; Tune, J.D. Role of potassium channels in coronary vasodilation. Exp. Biol. Med. 2010, 235, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Kryman, J.P.; McMillin, P.J.; White, R.E.; Carrier, G.O. A novel transduction mechanism mediating dopamine-induced vascular relaxation: Opening of BKCa channels by cyclic AMP-induced stimulation of the cyclic GMP-dependent protein kinase. J. Cardiovasc. Pharmacol. 1999, 34, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Barlow, R.S.; White, R.E. Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. Am. J. Physiol. 1998, 275, H1283–H1289. [Google Scholar] [PubMed]
- Barlow, R.S.; El-Mowafy, A.M.; White, R.E. H2O2 opens BKCa channels via the PLA2-arachidonic acid signaling cascade in coronary artery smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H475–H483. [Google Scholar] [CrossRef] [PubMed]
- Thengchaisri, N.; Kuo, L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: Role of cyclooxygenase and potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2255–H2263. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Hirata, Y.; Tokumura, A.; Nakaya, Y.; Fukuzawa, K. Protein kinase c-independent inhibition of the Ca2+-activated K+ channel by angiotensin II and endothelin-1. Biochem. Pharmacol. 1995, 49, 1051–1056. [Google Scholar] [CrossRef]
- Toro, L.; Amador, M.; Stefani, E. ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. Am. J. Physiol. 1990, 258, H912–H915. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.L.; Kim, H.S.; Jeng, A.Y. Dual action of endothelin-1 on the Ca2+-activated K+ channel in smooth muscle cells of porcine coronary artery. Eur. J. Pharmacol. 1991, 194, 31–36. [Google Scholar] [CrossRef]
- Scornik, F.S.; Toro, L. U46619, a thromboxane A2 agonist, inhibits KCa channel activity from pig coronary artery. Am. J. Physiol. 1992, 262, C708–C713. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.M.; Xia, Q.; Gao, Q.; Chen, M.; Wong, T.M. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J. Pharmacol. Exp. Ther. 2005, 312, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Sage, S.O.; van Breemen, C.; Cannell, M.B. Sodium calcium exchange in cultured bovine pulmonary artery endothelial cells. J. Physiol. 1991, 440, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.A.; Crea, F. Cardiopatia ischemica. In Medicina Interna Sistematica, 6th ed.; Edra-Masson; Elselvier Italia srl: Milan, Italy, 2010; Volume 1, pp. 103–149. ISBN 9788821439186. [Google Scholar]
- Wang, C.C.L.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Khaw, K.T.; Wareham, N.; Bingham, S.; Luben, R.; Welch, A.; Day, N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: The European prospective investigation into cancer in Norfolk. Ann. Intern. Med. 2004, 141, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Selvin, E.; Steffes, M.W.; Zhu, H.; Matsushita, K.; Wagenknecht, L.; Pankow, J.; Coresh, J.; Brancati, F.L. Glycated hemoglobin, diabetes and cardiovascular risk in nondiabetic adults. N. Engl. J. Med. 2010, 362, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Santos-Oliveira, R.; Purdy, C.; da Silva, M.P.; dos Anjos Carneiro-Leao, A.M.; Machado, M.; Einarson, T.R. Haemoglobin A1c levels and subsequent cardiovascular disease in persons without diabetes: A meta-analysis of prospective cohorts. Diabetologia 2011, 54, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Gu, K.; Cowie, C.C.; Harris, M.I. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971-1993. Diabetes Care 1998, 21, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Norhammar, A.; Lindbäck, J.; Rydén, L.; Wallentin, L.; Stenestrand, U. Improved but still high short- and long-term mortality rates after myocardial infarction in patients with diabetes mellitus: A time-trend report from the Swedish Register of Information and Knowledge about Swedish Heart Intensive Care Admission. Heart 2007, 93, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Constantino, M.I.; Molyneaux, L.; Limacher-Gisler, F.; Al-Saeed, A.; Luo, C.; Wu, T.; Twigg, S.M.; Yue, D.K.; Wong, J. Long-term complications and mortality in youngonset diabetes: Type 2 diabetes is more hazardous and lethal than type 1 diabetes. Diabetes Care 2013, 36, 3863–3869. [Google Scholar] [CrossRef] [PubMed]
- Soedamah-Muthu, S.S.; Fuller, J.H.; Mulnier, H.E.; Raleigh, V.S.; Lawrenson, R.A.; Colhoun, H.M. High risk of cardiovascular disease in patients with type 1 diabetes in the U.K.: A cohort study using the General Practice Research Database. Diabetes Care 2006, 29, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, K.; Kolodgie, F.D.; Lutter, C.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus Highlights. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Van Werkhoven, J.M.; Cademartiri, F.; Seitun, S.; Maffei, E.; Palumbo, A.; Martini, C.; Tarantini, G.; Kroft, L.J.; de Roos, A.; Weustink, A.C.; et al. Diabetes: Prognostic value of CT coronary angiography—Comparison with a nondiabetic population. Radiology 2010, 256, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Kolodgie, F.D.; Zieske, A.; Fowler, D.R.; Weber, D.K.; Varghese, P.J.; Farb, A.; Virmani, R. Morphologic findings of coronary atherosclerotic plaques in diabetics: A postmortem study. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.L.; Naya, M.; Foster, C.R.; Gaber, M.; Hainer, J.; Klein, J.; Dorbala, S.; Blankstein, R.; Di Carli, M.F. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012, 126, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Picchi, A.; Capobianco, S.; Qiu, T.; Focardi, M.; Zou, X.; Cao, J.M.; Zhang, C. Coronary microvascular dysfunction in diabetes mellitus: A review. World J. Cardiol. 2010, 2, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Nacci, C.; Tarquinio, M.; Montagnani, M. Molecular and clinical aspects of endothelial dysfunction in diabetes. Intern. Emerg. Med. 2009, 4, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Heart in diabetes: A microvascular disease. Diabetes Care 2011, 34, S145–S149. [Google Scholar] [CrossRef] [PubMed]
- Di Carli, M.F.; Janisse, J.; Ager, J.; Grunberger, G. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J. Am. Coll. Cardiol. 2003, 41, 1387–1393. [Google Scholar] [CrossRef]
- Yokoyama, I.; Momomura, S.I.; Ohtake, T.; Yonekura, K.; Nishikawa, J.; Sasaki, Y.; Omata, M.; et al. Reduced myocardial flow reserve in non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1997, 30, 1472–1477. [Google Scholar] [CrossRef]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial cell death in human diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Kotha, S.R.; Malireddy, S.; Selvaraju, V.; Satoskar, A.R.; Palesty, A.; McFadden, D.W.; Parinandi, N.L.; Maulik, N. Conundrum of pathogenesis of diabetic cardiomyopathy: Role of vascular endothelial dysfunction, reactive oxygen species, and mitochondria. Mol. Cell. Biochem. 2014, 386, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Di Carli, M.F.; Bianco-Batlles, D.; Landa, M.E.; Kazmers, A.; Groehn, H.; Muzik, O.; Grunberger, G. Effects of autonomic neuropathy on coronary blood flow in patients with diabetes mellitus. Circulation 1999, 100, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Taddei, S. Endothelial dysfunction in resistance arteries of hypertensive humans: Old and new conspirators. J. Cardiovasc. Pharmacol. 2016, 67, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Bennett, M. Aging and Atherosclerosis: Mechanisms, Functional Consequences, and Potential Therapeutics for Cellular Senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Morita, I.; Kaji, K.A.; Ikeda, M.; Nagao, M.; Murota, S. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem. Biophys. Res. Commun. 1993, 195, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [PubMed]
- Khaidakov, M.; Wang, X.; Mehta, J.L. Potential involvement of LOX-1 in functional consequences of endothelial senescence. PLoS ONE 2011, 6, e20964. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Meredith, A.; Lin, D.; McManus, B.M. The biological role of inflammation in atherosclerosis. Can. J. Cardiol. 2012, 28, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Balanescu, S.; Calmac, L.; Constantinescu, D.; Marinescu, M.; Onut, R.; Dorobantu, M. Systemic inflammation and early atheroma formation: Are they related? Maedica 2010, 5, 292–301. [Google Scholar] [PubMed]
- Farzaneh-Far, A.; Roman, M.J.; Lockshin, M.D.; Devereux, R.B.; Paget, S.A.; Crow, M.K.; Davis, A.; Sammaritano, L.; Levine, D.M.; Salmon, J.E. Relationship of antiphospholipid antibodies to cardiovascular manifestations of systemic lupus erythematosus. Arthritis Rheumatol. 2006, 54, 3918–3925. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Maggi, F.M.; Tragni, E. Low density lipoprotein oxidation, antioxidants, and atherosclerosis. Curr. Opin. Cardiol. 2000, 15, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular plat-form triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [PubMed]
- Grisar, J.; Aletaha, D.; Steiner, C.W.; Kapral, T.; Steiner, S.; Seidinger, D.; Weigel, G.; Schwarzinger, I.; Wolozcszuk, W.; Steiner, G.; et al. Depletion of endothelial progenitor cells in the peripheral blood of patients with rheumatoid arthritis. Circulation 2005, 111, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Li, W.; Chen, H.; Liu, H.; Huang, H.; Li, H. Advanced Glycation End Products Impair Voltage-Gated K+ Channels-Mediated Coronary Vasodilation in Diabetic Rats. PLoS ONE 2015, 10, e0142865. [Google Scholar] [CrossRef] [PubMed]
- Elvira, B.; Warsi, J.; Munoz, C.; Lang, F. SPAK and OSR1 Sensitivity of Voltage-Gated K+ Channel Kv1.5. J. Membr. Biol. 2015, 248, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Chapalamadugu, K.C.; Panguluri, S.K.; Bennett, E.S.; Kolliputi, N.; Tipparaju, S.M. High level of oxygen treatment causes cardiotoxicity with arrhythmias and redox modulation. Toxicol. Appl. Pharmacol. 2015, 282, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, S.; Niwano, S.; Niwano, H.; Murakami, M.; Ishikawa, S.; Masaki, Y.; Tamaki, H.; Toda, T.; Noda, Y.; Shimizu, T.; et al. Cardiomyocyte-derived mitochondrial superoxide causes myocardial electrical remodeling by downregulating potassium channels and related molecules. Circ. J. 2014, 78, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Wang, H.; Chai, Q.; Wang, X.; Shen, W.K.; Willis, M.S.; Lee, H.C.; Lu, T. Regulation of large conductance Ca2+-activated K+ (BK) channel β1 subunit expression by muscle RING finger protein 1 in diabetic vessels. J. Biol. Chem. 2014, 289, 10853–10864. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.-L.; Sun, X.; Chai, Q.; Li, J.; Thompson, B.; Shen, W.K.; Lu, T.; Lee, H.C. Regulation of vascular large-conductance calcium-activated potassium channels by Nrf2 signalling. Diabetes Vasc. Dis. Res. 2017, 14, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xie, A.; Singh, A.K.; Ehsan, A.; Choudhary, G.; Dudley, S.; Sellke, F.W.; Feng, J. Inactivation of Endothelial Small/Intermediate Conductance of Calcium-Activated Potassium Channels Contributes to Coronary Arteriolar Dysfunction in Diabetic Patients. J. Am. Heart Assoc. 2015, 4, e002062. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Jiang, B.; Wang, X.-L.; Lee, H.-C. Coronary arterial BK channel dysfunction exacerbates ischemia/reperfusion-induced myocardial injury in diabetic mice. Appl. Physiol. Nutr. Metab. 2016, 41, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Gonoi, T.; Clement, J.P., IV; Namba, N.; Inazawa, J.; Gonzalez, G.; Aguilar-Bryan, L.; Seino, S.; Bryan, J. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science 1995, 270, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. KATP channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Hu, H.; Deng, D.; Chen, M.; Xu, Z.; Wang, Y. Prediabetes is associated with genetic variations in the gene encoding the Kir6.2 subunit of the pancreatic ATP-sensitive potassium channel (KCNJ11): A case-control study in a Han Chinese youth population. J. Diabetes 2018, 10, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Sakura, H.; Wat, N.; Horton, V.; Millns, H.; Turner, R.C.; Ashcroft, F.M. Sequence variations in the human Kir6.2 gene, a subunit of the β-cell ATP-sensitive K-channel: No association with NIDDM in while Caucasian subjects or evidence of abnormal function when expressed in vitro. Diabetologia 1996, 39, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Tschritter, O.; Stumvoll, M.; Machicao, F.; Holzwarth, M.; Weisser, M.; Maerker, E.; Teigeler, A.; Häring, H.; Fritsche, A. The prevalent Glu23Lys polymorphism in the potassium inward rectifier 6.2 (KIR6.2) gene is associated with impaired glucagon suppression in response to hyperglycemia. Diabetes 2002, 51, 2854–2860. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.C.; Marshall, B.A.; Ensor, N.E.A.; Corbett, J.A.; Nichols, C.G. Targeted overactivity of β cell KATP channels induces profound neonatal diabetes. Cell 2000, 100, 645–654. [Google Scholar] [CrossRef]
- Girard, C.A.; Wunderlich, F.T.; Shimomura, K.; Collins, S.; Kaizik, S.; Proks, P.; Abdulkader, F.; Clark, A.; Ball, V.; Zubcevic, L.; et al. Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic β cells recapitulates neonatal diabetes. J. Clin. Investig. 2009, 119, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Remedi, M.S.; Kurata, H.T.; Scott, A.; Wunderlich, F.T.; Rother, E.; Kleinridders, A.; Tong, A.; Brüning, J.C.; Koster, J.C.; Nichols, C.G. Secondary consequences of β cell inexcitability: Identification and prevention in a murine model of KATP-induced neonatal diabetes mellitus. Cell Metab. 2009, 9, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [PubMed]
- Gylfe, E.; Gilon, P. Glucose regulation of glucagon secretion. Diabetes Res. Clin. Pract. 2014, 103, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rastegari, A.; Rabbani, M.; Sadeghi, H.M.; Imani, E.F.; Hasanzadeh, A.; Moazen, F. Association of KCNJ11 (E23K) gene polymorphism with susceptibility to type 2 diabetes in Iranian patients. Adv. Biomed. Res. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Na, R.; Xu, R.; Wang, S.; Sheng, H.; Wu, W.; Qu, Y. Quantitative assessment of the effect of KCNJ11 gene polymorphism on the risk of type 2 diabetes. PLoS ONE 2014, 9, e93961. [Google Scholar] [CrossRef] [PubMed]
- Souza, S.W.; Alcazar, L.P.; Arakaki, P.A.; Santos-Weiss, I.C.R.; Alberton, D.; Picheth, G.; Rego, F.G.M. Polymorphism E23K (rs5219) in the KCNJ11 gene in Euro-Brazilian subjects with type 1 and 2 diabetes. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.J.; Lv, Y.; Huang, Q.Y. Meta-analysis of association of common variants in the KCNJ11-ABCC8 region with type 2 diabetes. Genet. Mol. Res. 2013, 12, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Emdin, C.A.; Klarin, D.; Natarajan, P.; Florez, J.C.; Kathiresan, S.; Khera, A.V. Genetic Variation at the Sulfonylurea Receptor, Type 2 Diabetes, and Coronary Heart Disease. Diabetes 2017, 66, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.C.; Cull, C.A.; Frighi, V.; Holman, R.R. UK Prospective Diabetes Study (UKPDS) Group. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: Progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 1999, 281, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Förstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Barakat, A.; Lieu, D. Differential responsiveness of vascular endothelial cells to different types of fluid mechanical shear stress. Cell Biochem. Biophys. 2003, 38, 323–343. [Google Scholar] [CrossRef]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Kaazempur-Mofrad, M.R.; Natarajan, S.; Zhang, Y.; Vaughn, S.; Blackman, B.R.; Kamm, R.D.; García-Cardeña, G.; Gimbrone, M.A. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Passerini, A.G.; Polacek, D.C.; Shi, C.; Francesco, N.M.; Manduchi, E.; Grant, G.R.; Pritchard, W.F.; Powell, S.; Chang, G.Y.; Stoeckert, C.J. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc. Natl. Acad. Sci. USA 2004, 101, 2482–2487. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Anti-inflammatory mechanisms in the vascular wall. Circ. Res. 2001, 88, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Gojova, A.; Barakat, A.I. Flow-Activated Ion Channels in Vascular Endothelium. Cell Biochem. Biophys. 2006, 46, 277–284. [Google Scholar] [CrossRef]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, A.C.; Gangopadhyay, N.N.; Devor, D.C. Kinase-dependent regulation of the intermediate conductance, calcium-dependent potassium channel, hIK1. J. Biol. Chem. 2000, 275, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Li, Z.; Ko, K.; Choudhury, P.; Albaqumi, M.; Johnson, A.K.; Yan, Y.; Backer, J.M.; Unutmaz, D.; Coetzee, W.A.; et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell. 2006, 24, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Neylon, C.B.; Lang, R.J.; Fu, Y.; Bobik, A.; Reinhart, P.H. Molecular cloning, characterizationoftheintermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: Relationship between KCa channel diversity and smooth muscle cell function. Circ. Res. 1999, 85, e3343. [Google Scholar] [CrossRef]
- Chung, I.; Zelivyanskaya, M.; Gendelman, H.E. Mononuclear phagocyte biophysiology influences brain transendothelial and tissue migration: Implication for HIV-1-associated dementia. J. Neuroimmunol. 2002, 122, 40–54. [Google Scholar] [CrossRef]
- Tharp, D.L.; Wamhoff, B.R.; Turk, J.R.; Bowles, D.K. Upregulation of intermediate-conductance Ca2+-activated K+ channel (IKCa1) mediates phenotypic modulation of coronary smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2493–H2503. [Google Scholar] [CrossRef] [PubMed]
- Wolfs, J.L.; Wielders, S.J.; Comfurius, P.; Lindhout, T.; Giddings, J.C.; Zwaal, R.F.; Bevers, E.M. Reversible inhibition of the platelet procoagulant response through manipulation of the Gardos channel. Blood 2006, 108, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Liu, M.; Liu, C. Exenatide Inhibits the KCa3.1 Channels of Aortic Vascular Smooth Muscle in Diabetic Rats. Acta Cardiol. Sin. 2017, 33, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.; Lemaitre, V.; Takai, J.; Miura, H. The intermediate conductance Ca2+-activated K+ channel KCa3.1 regulates proliferation of human coronary smooth muscle cells. Circulation 2012, 126, A16586. [Google Scholar]
- Su, X.L.; Zhang, H.; Yu, W.; Wang, S.; Zhu, W.J. Role of KCa3.1 channels in proliferation and migration of vascular smooth muscle cells by diabetic rat serum. Chin. J. Physiol. 2013, 56, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wang, Y.; Zhang, W.; Zhao, L.M.; Li, G.R.; Deng, X.L. Insulin-mediated upregulation of K3.1 channels promotes cell migration and proliferation in rat vascular smooth muscle. J. Mol. Cell. Cardiol. 2011, 51, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.M.; Su, X.L.; Wang, Y.; Li, G.R.; Deng, X.L. KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation end products in cultured rat vascular smooth muscle cells. Lab. Investig. 2013, 93, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Li, C.; Wu, Y.; Shen, L.; Ma, J.; Qian, J.; Ge, J. Role of KCa3.1 Channels in Macrophage Polarization and Its Relevance in Atherosclerotic Plaque Instability. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Bernheim, L.; Bader, C.R. Human myoblast differentiation: Ca2+ channels are activated by K+ channels. News Physiol. Sci. 2002, 17, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, X.-J.; Wang, Y.-F.; Wang, D.Q.; Yuan, Z.Y. Role of Kir2.1 in human monocyte-derived foam cell maturation. J. Cell. Mol. Med. 2016, 20, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Cheong, A.; Li, J.; Sukumar, P.; Kumar, B.; Zeng, F.; Riches, K.; Munsch, C.; Wood, I.C.; Porter, K.E.; Beech, D.J. Potent suppression of vascular smooth muscle cell migration and human neointimal hyperplasia by KV1.3 channel blockers. Cardiovasc. Res. 2011, 89, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Kan, X.-H.; Gao, H.-Q.; Ma, Z.-Y.; Liu, L.; Ling, M.Y.; Wang, Y.Y. Kv1.3 potassium channel mediates macrophage migration in atherosclerosis by regulating ERK activity. Arch. Biochem. Biophys. 2016, 591, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Calabresi, P.A.; Allie, R.; Yun, S.; Pennington, M.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J. Clin. Investig. 2003, 111, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, N.; David, M.; Bielańska, J.; González, T.; Parra, D.; Soler, C.; Comes, N.; Valenzuela, C.; Felipe, A. Immunomodulatory effects of diclofenac in leukocytes through the targeting of Kv1.3 voltage-dependent potassium channels. Biochem. Pharmacol. 2010, 80, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Kazama, I.; Tamada, T.; Tachi, M. Usefulness of targeting lymphocyte Kv1.3-channels in the treatment of respiratory diseases. Inflamm. Res. 2015, 64, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.-Y.; Ma, Z.-Y.; Wang, Y.-Y.; Qi, J.; Liu, L.; Li, L.; Zhang, Y. Up-regulated ATP-sensitive potassium channels play a role in increased inflammation and plaque vulnerability in macrophages. Atherosclerosis 2013, 226, 348–355. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | DM (Tot. 67) | NDM (Tot. 175) | p Value |
|---|---|---|---|
| Gender [male] % (n) | 68.6 (46) | 64.5 (113) | NS |
| Smoke habit % (n) | 34.3 (23) | 41.1 (72) | NS |
| Hypertension % (n) | 86.5 (58) | 63.4 (111) | 0.0002 |
| Dyslipidemia % (n) | 88 (59) | 64 (112) | 0.0002 |
| Family history of CVD % (n) | 80.5 (54) | 70.2 (123) | NS |
| ACS % (n) | 58.2 (39) | 51.4 (90) | NS |
| CAD % (n) | 70.1 (47) | 61.7 (108) | NS |
| CMD % (n) | 8.9 (6) | 22.8 (40) | 0.016 |
| NORM % (n) | 20.8 (14) | 15.3 (27) | NS |
| Ion Channel | Protein | Gene | SNPs | Effect | References |
|---|---|---|---|---|---|
| K-ATP | Kir6.2 | KCNJ11 | rs5210 |
| [95] |
| K-ATP | Kir6.2 | KCNJ11 | rs5215_GG |
| [11] |
| K-ATP | Kir6.2 | KCNJ11 | rs5218_CT |
| [11] |
| K-ATP | Kir6.2 | KCNJ11 | rs5219_AA; rs5219 (E23K-Glu23Lys) |
| [11,84,85,86,92,93,94] |
| K-ATP | SUR1 | ABCC8 | rs757110 |
| [95] |
| K-ATP | SUR1 | ABCC8 | p.A1369S |
| [96,97] |
| K-ATP | SUR1 | ABCC8 | Ser1369Ala |
| [84,85] |
| Nav | Nav1.5 | SCN5A | rs1805124_GG |
| [11] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; De Marchis, M.; Palmirotta, R.; Volterrani, M.; Mancone, M.; Fedele, F. Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels. Int. J. Mol. Sci. 2018, 19, 802. https://doi.org/10.3390/ijms19030802
Severino P, D’Amato A, Netti L, Pucci M, De Marchis M, Palmirotta R, Volterrani M, Mancone M, Fedele F. Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels. International Journal of Molecular Sciences. 2018; 19(3):802. https://doi.org/10.3390/ijms19030802
Chicago/Turabian StyleSeverino, Paolo, Andrea D’Amato, Lucrezia Netti, Mariateresa Pucci, Marialaura De Marchis, Raffaele Palmirotta, Maurizio Volterrani, Massimo Mancone, and Francesco Fedele. 2018. "Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels" International Journal of Molecular Sciences 19, no. 3: 802. https://doi.org/10.3390/ijms19030802
APA StyleSeverino, P., D’Amato, A., Netti, L., Pucci, M., De Marchis, M., Palmirotta, R., Volterrani, M., Mancone, M., & Fedele, F. (2018). Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels. International Journal of Molecular Sciences, 19(3), 802. https://doi.org/10.3390/ijms19030802