Regulation of T-Cell Signaling by Post-Translational Modifications in Autoimmune Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. T-Cell Signal Transduction Pathways
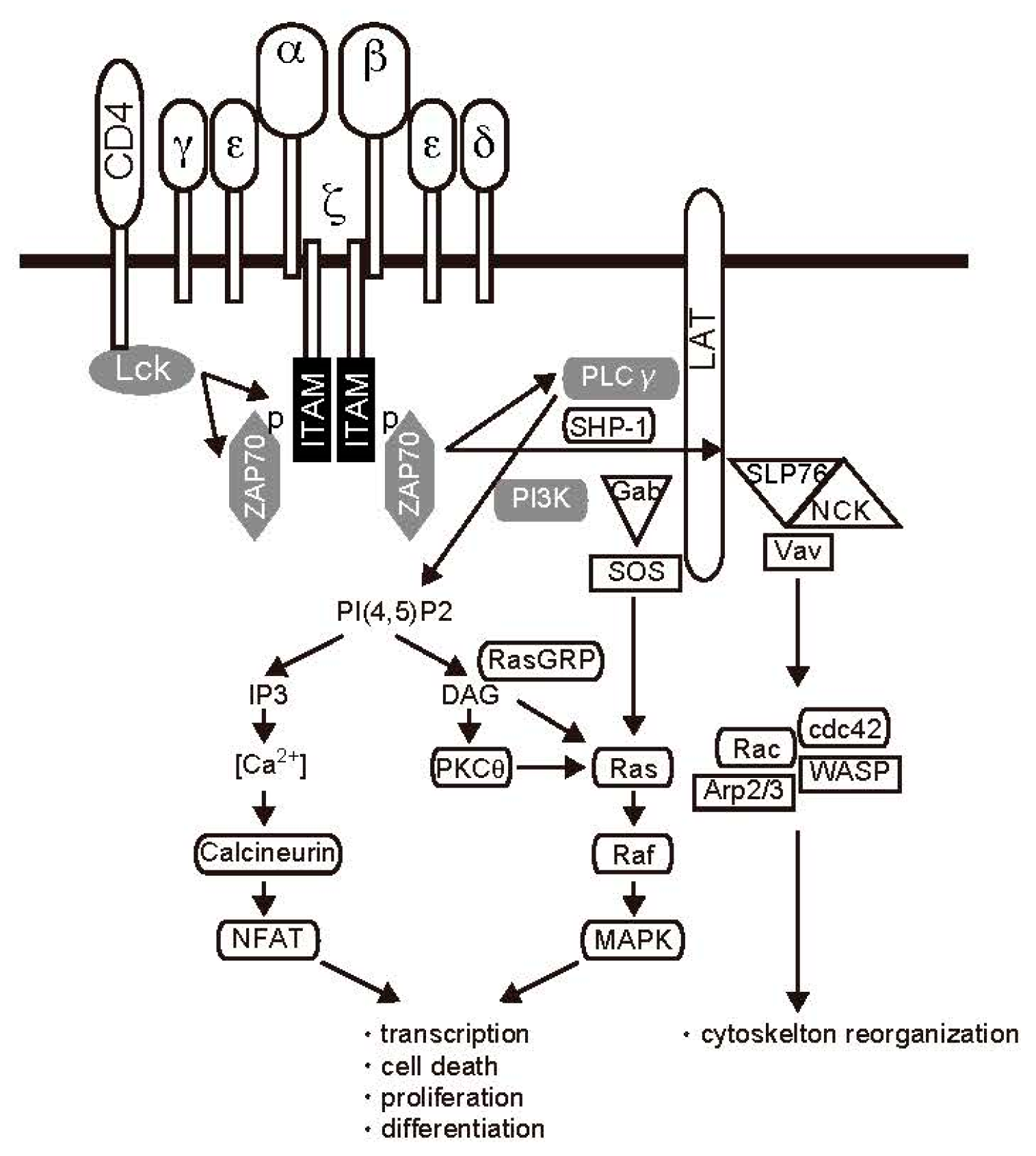
2.1. TCR Signal Transduction
2.2. Cytokine Receptor Signaling
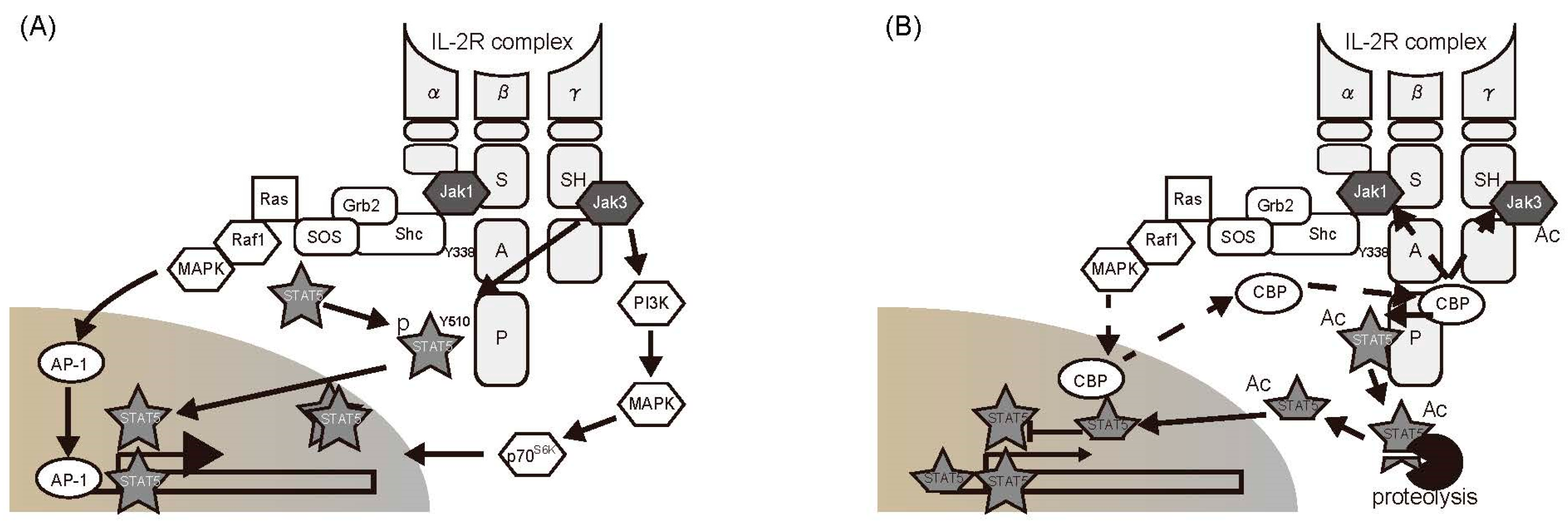
2.3. The Janus Kinase (JAK)/STAT Pathway
2.4. The Ras/MAPK Pathway
2.5. The Phosphatidylinositol 3-Kinase (PI3K)/Akt Pathway
3. Signaling Regulation and Autoimmune Disease
3.1. Negative Regulation by Protein Tyrosine Phosphatases (PTPs)
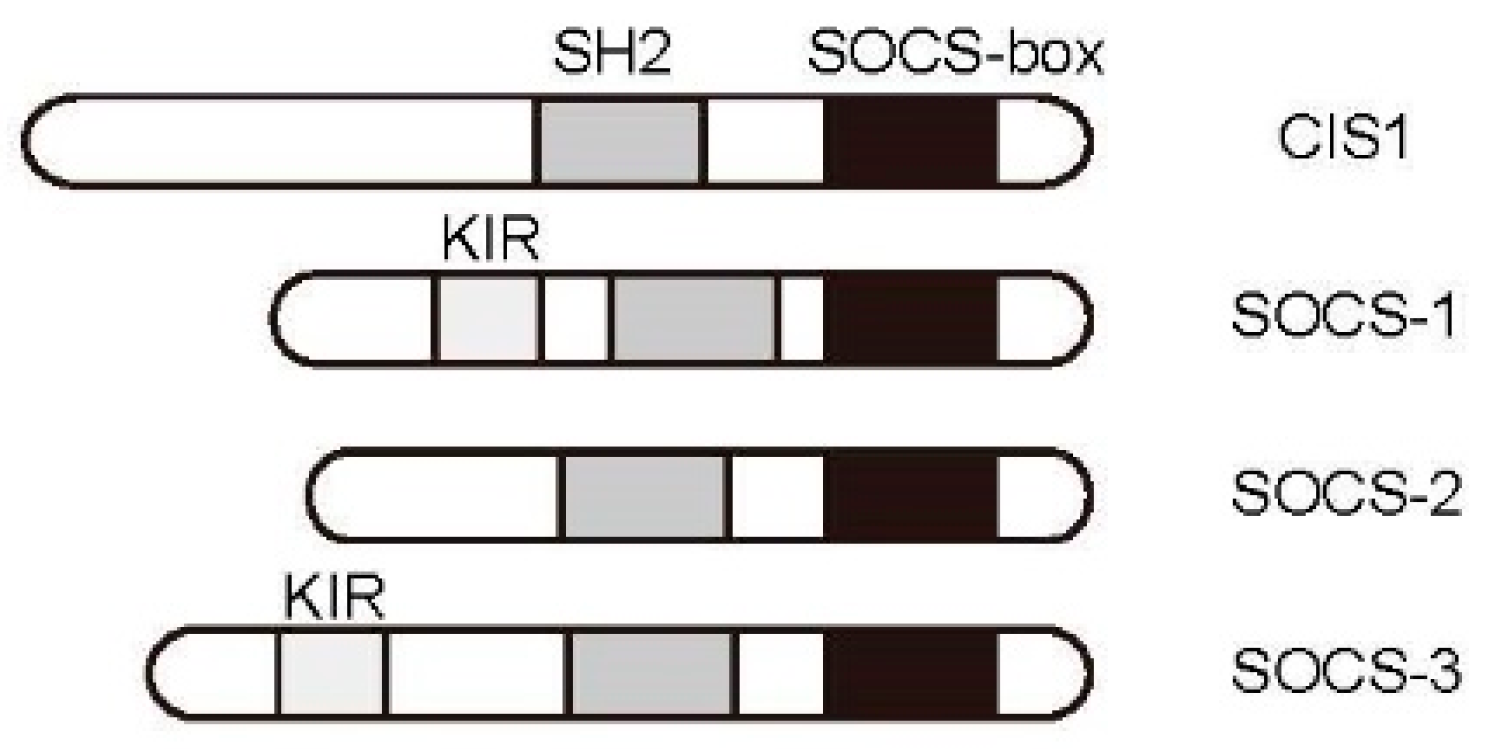
3.2. JAK/STAT Pathway Regulation by the Cytokine-Induced SH2-Containing Protein (CIS)/Suppressor of Cytokine Signaling (SOCS) Family
3.3. Regulation of TCR Signaling and Associated Diseases
3.4. T Cell-Targeted Nanomedicine
4. T-Cell Signaling Inhibitors and Autoimmune Diseases
5. T-Cell Function and Cytoplasmic Acetylation
5.1. Microtubule Regulation by Cytoplasmic Acetylation and T-Cell Function
5.2. Regulation of Cytokine Cascades by Acetylation of Signaling Molecules in the Cytoplasm
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| APCs | antigen-presenting cells |
| CD | cluster of differentiation |
| CIS | cytokine-induced SH2-containing protein |
| CREB | cyclic AMPresponsive element binding protein |
| DAG | diacylglycerol |
| ERK | extracellular signal-regulated kinase |
| IFN | interferon |
| IL | interleukin |
| IP3 | inositol 1,4,5-triphosphat |
| ITAM | immune-receptor tyrosine-based activation motif |
| ITIM | immuno-receptor tyrosine-based inhibitory motif |
| JAK | Janus kinase |
| Lck | lymphocyte-specific protein tyrosine kinase |
| MAPK | mitogen-activated protein kinase |
| MEK | mitogen-activated protein/extracellular signal-regulated kinase |
| MHC | major histocompatibility complex |
| NEMO | NF-κB essential modulator |
| NFAT | nuclear factor of activated T cells |
| NF-κB | nuclear factor-κB |
| PIP2 | phosphatidylinositol-4,5-diphosphate |
| PI3K | phosphatidylinositol 3-kinase |
| PKCθ | protein kinase Cθ |
| PLCγ | phospholipase Cγ |
| PTP | protein tyrosine phosphatase |
| RA | rheumatoid arthritis |
| Ras | rat sarcoma |
| SOCS | suppressor of cytokine signaling |
| Stat | signal transducers and activators of transcription |
| TCR | T-cell receptor |
| ZAP70 | ζ chain-associated 70 kDa tyrosine phosphoprotein |
References
- Gasteiger, G.; Ataide, M.; Kastenmuller, W. Lymph node—An organ for T-cell activation and pathogen defense. Immunol. Rev. 2016, 271, 200–220. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The cellular context of T cell signaling. Immunity 2009, 30, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, D. T cell antigen receptor signal transduction pathways. Annu. Rev. Immunol. 1996, 14, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Aramburu, J.; Garcia-Rodriguez, C.; Viola, J.P.; Raghavan, A.; Tahiliani, M.; Zhang, X.; Qin, J.; Hogan, P.G.; Rao, A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 2000, 6, 539–550. [Google Scholar] [CrossRef]
- Kuwabara, T.; Kasai, H.; Kondo, M. Acetylation Modulates IL-2 Receptor Signaling in T Cells. J. Immunol. 2016, 197, 4334–4343. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.; Knoechel, B.; Nagabhushanam, V.; Abbas, A.K. T-cell tolerance and autoimmunity to systemic and tissue-restricted self-antigens. Immunol. Rev. 2005, 204, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Fathman, C.G.; Lineberry, N.B. Molecular mechanisms of CD4+ T-cell anergy. Nat. Rev. Immunol. 2007, 7, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Lo, B.; Abdel-Motal, U.M. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr. Opin. Immunol. 2017, 49, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Alarcon, B.; Wileman, T.; Terhorst, C. The T cell receptor/CD3 complex: A dynamic protein ensemble. Annu. Rev. Immunol. 1988, 6, 629–662. [Google Scholar] [CrossRef] [PubMed]
- Hermiston, M.L.; Xu, Z.; Majeti, R.; Weiss, A. Reciprocal regulation of lymphocyte activation by tyrosine kinases and phosphatases. J. Clin. Investig. 2002, 109, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M. A new trigger for T cells. Cell 2002, 110, 285–287. [Google Scholar] [CrossRef]
- Gong, Q.; Cheng, A.M.; Akk, A.M.; Alberola-Ila, J.; Gong, G.; Pawson, T.; Chan, A.C. Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nat. Immunol. 2001, 2, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Weiss, A. T cell receptor signalling. J. Cell Sci. 2001, 114, 243–244. [Google Scholar] [PubMed]
- Hara, H.; Wada, T.; Bakal, C.; Kozieradzki, I.; Suzuki, S.; Suzuki, N.; Nghiem, M.; Griffiths, E.K.; Krawczyk, C.; Bauer, B.; et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity 2003, 18, 763–775. [Google Scholar] [CrossRef]
- Pomerantz, J.L.; Denny, E.M.; Baltimore, D. CARD11 mediates factor-specific activation of NF-κB by the T cell receptor complex. EMBO J. 2002, 21, 5184–5194. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; You, Y.; Case, S.M.; McAllister-Lucas, L.M.; Wang, L.; DiStefano, P.S.; Nunez, G.; Bertin, J.; Lin, X. A requirement for CARMA1 in TCR-induced NF-κB activation. Nat. Immunol. 2002, 3, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Oh-hora, M.; Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 2008, 20, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J.; Pozzan, T. The endoplasmic reticulum Ca2+ store: A view from the lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Liu, J.O. Calmodulin-dependent phosphatase, kinases, and transcriptional corepressors involved in T-cell activation. Immunol. Rev. 2009, 228, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, C.R.; Parge, H.E.; Knighton, D.R.; Lewis, C.T.; Pelletier, L.A.; Tempczyk, A.; Kalish, V.J.; Tucker, K.D.; Showalter, R.E.; Moomaw, E.W.; et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature 1995, 378, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Ouyang, W.; Farrar, J.D.; Yang, J.; Ranganath, S.; Asnagli, H.; Afkarian, M.; Murphy, T.L. Signaling and transcription in T helper development. Annu. Rev. Immunol. 2000, 18, 451–494. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef] [PubMed]
- De Becker, G.; Moulin, V.; Tielemans, F.; De Mattia, F.; Urbain, J.; Leo, O.; Moser, M. Regulation of T helper cell differentiation in vivo by soluble and membrane proteins provided by antigen-presenting cells. Eur. J. Immunol. 1998, 28, 3161–3171. [Google Scholar] [CrossRef]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef]
- Maldonado-Lopez, R.; De Smedt, T.; Michel, P.; Godfroid, J.; Pajak, B.; Heirman, C.; Thielemans, K.; Leo, O.; Urbain, J.; Moser, M. CD8alpha+ and CD8alpha− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 1999, 189, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.F. A novel family of growth factor receptors: A common binding domain in the growth hormone, prolactin, erythropoietin and IL-6 receptors, and the p75 IL-2 receptor β-chain. Biochem. Biophys. Res. Commun. 1989, 164, 788–795. [Google Scholar] [CrossRef]
- Miyajima, A.; Kitamura, T.; Harada, N.; Yokota, T.; Arai, K. Cytokine receptors and signal transduction. Annu. Rev. Immunol. 1992, 10, 295–331. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Kishimoto, T. Signaling mechanisms through cytokine receptors that share signal transducing receptor components. Curr. Opin. Immunol. 1995, 7, 17–23. [Google Scholar] [CrossRef]
- Sugamura, K.; Asao, H.; Kondo, M.; Tanaka, N.; Ishii, N.; Ohbo, K.; Nakamura, M.; Takeshita, T. The interleukin-2 receptor gamma chain: Its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu. Rev. Immunol. 1996, 14, 179–205. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon α/β signaling pathway. Cell 1992, 70, 313–322. [Google Scholar] [CrossRef]
- Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Yi, T.; Tang, B.; Miura, O.; Ihle, J.N. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 1993, 74, 227–236. [Google Scholar] [CrossRef]
- Ihle, J.N.; Witthuhn, B.A.; Quelle, F.W.; Yamamoto, K.; Silvennoinen, O. Signaling through the hematopoietic cytokine receptors. Annu. Rev. Immunol. 1995, 13, 369–398. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Sato, N.; Sakamaki, K.; Terada, N.; Arai, K.; Miyajima, A. Signal transduction by the high-affinity GM-CSF receptor: Two distinct cytoplasmic regions of the common β subunit responsible for different signaling. EMBO J. 1993, 12, 4181–4189. [Google Scholar] [PubMed]
- Blaikie, P.; Immanuel, D.; Wu, J.; Li, N.; Yajnik, V.; Margolis, B. A region in Shc distinct from the SH2 domain can bind tyrosine-phosphorylated growth factor receptors. J. Biol. Chem. 1994, 269, 32031–32034. [Google Scholar] [PubMed]
- Lioubin, M.N.; Algate, P.A.; Tsai, S.; Carlberg, K.; Aebersold, A.; Rohrschneider, L.R. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 1996, 10, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; Lin, J.X. Cytokine receptor signaling pathways. J. Allergy Clin. Immunol. 2000, 105, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nakafuku, M.; Miyajima, A.; Kaziro, Y. Involvement of ras p21 protein in signal-transduction pathways from interleukin 2, interleukin 3, and granulocyte/macrophage colony-stimulating factor, but not from interleukin 4. Proc. Natl. Acad. Sci. USA 1991, 88, 3314–3318. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Muto, A.; Watanabe, S.; Miyajima, A.; Yokota, T.; Arai, K. Granulocyte-macrophage colony-stimulating factor provokes RAS activation and transcription of c-fos through different modes of signaling. J. Biol. Chem. 1996, 271, 7587–7592. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Chen, H.E.; Goelz, S.; Larner, A.C.; Neel, B.G. Differential regulation of the α/β interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 1995, 15, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Petricoin, E., III; Benjamin, C.; Pine, R.; Weber, M.J.; Larner, A.C. Requirement for MAP kinase (ERK2) activity in interferon α- and interferon β-stimulated gene expression through STAT proteins. Science 1995, 269, 1721–1723. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Ihle, J.N. STATs and MAPKs: Obligate or opportunistic partners in signaling. Bioessays 1996, 18, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Abell, K.; Watson, C.J. The Jak/Stat pathway: A novel way to regulate PI3K activity. Cell Cycle 2005, 4, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.H.; Charbonneau, H.; Tonks, N.K. Protein tyrosine phosphatases: A diverse family of intracellular and transmembrane enzymes. Science 1991, 253, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Pallen, C.J.; Tan, Y.H.; Guy, G.R. Protein phosphatases in cell signaling. Curr. Opin. Cell Biol. 1992, 4, 1000–1007. [Google Scholar] [CrossRef]
- Gauzzi, M.C.; Velazquez, L.; McKendry, R.; Mogensen, K.E.; Fellous, M.; Pellegrini, S. Interferon-α-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J. Biol. Chem. 1996, 271, 20494–20500. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Witthuhn, B.A.; Matsuda, T.; Kohlhuber, F.; Kerr, I.M.; Ihle, J.N. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol. Cell. Biol. 1997, 17, 2497–2501. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Schlessinger, J. Switching signals on or off by receptor dimerization. Cell 1998, 94, 277–280. [Google Scholar] [CrossRef]
- Jiao, H.; Berrada, K.; Yang, W.; Tabrizi, M.; Platanias, L.C.; Yi, T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol. Cell. Biol. 1996, 16, 6985–6992. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.J.; Harbor, P.; Tabrizi, M.; Yi, T.; Williams, B.R. Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4- and IL-13-dependent signal transduction. J. Biol. Chem. 1998, 273, 33893–33896. [Google Scholar] [CrossRef] [PubMed]
- Migone, T.S.; Cacalano, N.A.; Taylor, N.; Yi, T.; Waldmann, T.A.; Johnston, J.A. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3845–3850. [Google Scholar] [CrossRef] [PubMed]
- Kamata, T.; Yamashita, M.; Kimura, M.; Murata, K.; Inami, M.; Shimizu, C.; Sugaya, K.; Wang, C.R.; Taniguchi, M.; Nakayama, T. src homology 2 domain-containing tyrosine phosphatase SHP-1 controls the development of allergic airway inflammation. J. Clin. Investig. 2003, 111, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef]
- Qu, C.K.; Nguyen, S.; Chen, J.; Feng, G.S. Requirement of SHP-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood 2001, 97, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Shen, R.; Feng, G.S.; Yang, Y.C. Molecular characterization of specific interactions between SHP-2 phosphatase and JAK tyrosine kinases. J. Biol. Chem. 1997, 272, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Yu, D.H.; Feng, G.S. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell. Biol. 1999, 19, 2416–2424. [Google Scholar] [CrossRef] [PubMed]
- Klingmuller, U.; Lorenz, U.; Cantley, L.C.; Neel, B.G.; Lodish, H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 1995, 80, 729–738. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ohkubo, T.; Kiguchi, T.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Hara, T.; Miyajima, A. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995, 14, 2816–2826. [Google Scholar] [PubMed]
- Endo, T.A.; Masuhara, M.; Yokouchi, M.; Suzuki, R.; Sakamoto, H.; Mitsui, K.; Matsumoto, A.; Tanimura, S.; Ohtsubo, M.; Misawa, H.; et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997, 387, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.C.; Restifo, N.P. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009, 30, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Misawa, H.; Sakamoto, H.; Masuhara, M.; Sasaki, A.; Wakioka, T.; Ohtsuka, S.; Imaizumi, T.; Matsuda, T.; Ihle, J.N.; et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hanada, T.; Mitsuyama, K.; Yoshida, T.; Kamizono, S.; Hoshino, T.; Kubo, M.; Yamashita, A.; Okabe, M.; Takeda, K.; et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 2001, 93, 471–481. [Google Scholar] [CrossRef]
- Veenbergen, S.; Bennink, M.B.; de Hooge, A.S.; Arntz, O.J.; Smeets, R.L.; van den Berg, W.B.; van de Loo, F.A. Splenic suppressor of cytokine signaling 3 transgene expression affects T cell responses and prevents development of collagen-induced arthritis. Arthritis Rheum. 2008, 58, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Shouda, T.; Yoshida, T.; Hanada, T.; Wakioka, T.; Oishi, M.; Miyoshi, K.; Komiya, S.; Kosai, K.; Hanakawa, Y.; Hashimoto, K.; et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J. Clin. Investig. 2001, 108, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.D., Jr. Rheumatoid arthritis. Pathophysiology and implications for therapy. N. Engl. J. Med. 1990, 322, 1277–1289. [Google Scholar] [PubMed]
- Sakaguchi, N.; Takahashi, T.; Hata, H.; Nomura, T.; Tagami, T.; Yamazaki, S.; Sakihama, T.; Matsutani, T.; Negishi, I.; Nakatsuru, S.; et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 2003, 426, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Giese, B.; Roderburg, C.; Sommerauer, M.; Wortmann, S.B.; Metz, S.; Heinrich, P.C.; Muller-Newen, G. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J. Cell Sci. 2005, 118, 5129–5140. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Scheller, J.; Elson, G.; Jones, S.A. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J. Leukoc. Biol. 2006, 80, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Skoda, R.C.; Cardiff, R.D.; Campos-Torres, J.; Leder, P.; Ornitz, D.M. Expression of LIF in transgenic mice results in altered thymic epithelium and apparent interconversion of thymic and lymph node morphologies. EMBO J. 1994, 13, 1375–1385. [Google Scholar] [PubMed]
- Metcalfe, S.M.; Watson, T.J.; Shurey, S.; Adams, E.; Green, C.J. Leukemia inhibitory factor is linked to regulatory transplantation tolerance. Transplantation 2005, 79, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Mahic, M.; Kalland, M.E.; Aandahl, E.M.; Torgersen, K.M.; Tasken, K. Human naturally occurring and adaptive regulatory T cells secrete high levels of leukaemia inhibitory factor upon activation. Scand. J. Immunol. 2008, 68, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Gao, W.; Whiston, R.; Strom, T.B.; Metcalfe, S.; Fahmy, T.M. Modulation of CD4+ T lymphocyte lineage outcomes with targeted, nanoparticle-mediated cytokine delivery. Mol. Pharm. 2011, 8, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Thompson, L.; Zhou, Q.; Putheti, P.; Fahmy, T.M.; Strom, T.B.; Metcalfe, S.M. Treg versus Th17 lymphocyte lineages are cross-regulated by LIF versus IL-6. Cell Cycle 2009, 8, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, S.M. LIF in the regulation of T-cell fate and as a potential therapeutic. Genes Immun. 2011, 12, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [PubMed]
- Patton, D.T.; Garden, O.A.; Pearce, W.P.; Clough, L.E.; Monk, C.R.; Leung, E.; Rowan, W.C.; Sancho, S.; Walker, L.S.; Vanhaesebroeck, B.; et al. Cutting edge: The phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 2006, 177, 6598–6602. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Camps, M.; Ji, H. PI3Kδ and PI3Kγ: Partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. 2007, 7, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Banham-Hall, E.; Clatworthy, M.R.; Okkenhaug, K. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Haruta, K.; Mori, S.; Tamura, N.; Sasaki, A.; Nagamine, M.; Yaguchi, S.; Kamachi, F.; Enami, J.; Kobayashi, S.; Yamori, T.; et al. Inhibitory effects of ZSTK474, a phosphatidylinositol 3-kinase inhibitor, on adjuvant-induced arthritis in rats. Inflamm. Res. 2012, 61, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.E.; Blumenkopf, T.A.; Brissette, W.H.; Brown, M.F.; Casavant, J.M.; Shang-Poa, C.; Doty, J.L.; Elliott, E.A.; Fisher, M.B.; Hines, M.; et al. Discovery of CP-690,550: A potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J. Med. Chem. 2010, 53, 8468–8484. [Google Scholar] [CrossRef] [PubMed]
- Milici, A.J.; Kudlacz, E.M.; Audoly, L.; Zwillich, S.; Changelian, P. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R14. [Google Scholar] [CrossRef] [PubMed]
- Park, H.B.; Oh, K.; Garmaa, N.; Seo, M.W.; Byoun, O.J.; Lee, H.Y.; Lee, D.S. CP-690550, a Janus kinase inhibitor, suppresses CD4+ T-cell-mediated acute graft-versus-host disease by inhibiting the interferon-γ pathway. Transplantation 2010, 90, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kimura, A.; Fukaya, T.; Sekiya, T.; Morita, R.; Shichita, T.; Inoue, H.; Yoshimura, A. Low dose CP-690,550 (tofacitinib), a pan-JAK inhibitor, accelerates the onset of experimental autoimmune encephalomyelitis by potentiating Th17 differentiation. Biochem. Biophys. Res. Commun. 2012, 418, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Kremer, J.; Cush, J.; Schulze-Koops, H.; Connell, C.A.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; Kanik, K.S.; et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Blanco, R.; Charles-Schoeman, C.; Wollenhaupt, J.; Zerbini, C.; Benda, B.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; Zwillich, S.H.; et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: A randomised phase 3 trial. Lancet 2013, 381, 451–460. [Google Scholar] [CrossRef]
- Kremer, J.; Li, Z.G.; Hall, S.; Fleischmann, R.; Genovese, M.; Martin-Mola, E.; Isaacs, J.D.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: A randomized trial. Ann. Intern. Med. 2013, 159, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Tanaka, Y.; Fleischmann, R.; Keystone, E.; Kremer, J.; Zerbini, C.; Cardiel, M.H.; Cohen, S.; Nash, P.; Song, Y.W.; et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: Twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013, 65, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Fleischmann, R.; Hall, S.; Wilkinson, B.; Bradley, J.D.; Gruben, D.; Koncz, T.; Krishnaswami, S.; Wallenstein, G.V.; Zang, C.; et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N. Engl. J. Med. 2014, 370, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Kremer, J.; Zamani, O.; Ludivico, C.; Krogulec, M.; Xie, L.; Beattie, S.D.; Koch, A.E.; Cardillo, T.E.; Rooney, T.P.; et al. Baricitinib in Patients with Refractory Rheumatoid Arthritis. N. Engl. J. Med. 2016, 374, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Dougados, M.; van der Heijde, D.; Chen, Y.C.; Greenwald, M.; Drescher, E.; Liu, J.; Beattie, S.; Witt, S.; de la Torre, I.; Gaich, C.; et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: Results from the RA-BUILD study. Ann. Rheum. Dis. 2017, 76, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Schiff, M.; van der Heijde, D.; Ramos-Remus, C.; Spindler, A.; Stanislav, M.; Zerbini, C.A.; Gurbuz, S.; Dickson, C.; de Bono, S.; et al. Baricitinib, Methotrexate, or Combination in Patients with Rheumatoid Arthritis and No or Limited Prior Disease-Modifying Antirheumatic Drug Treatment. Arthritis Rheumatol. 2017, 69, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamazaki, S.; Yamagami, K.; Kuno, M.; Morita, Y.; Okuma, K.; Nakamura, K.; Chida, N.; Inami, M.; Inoue, T.; et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J. Pharmacol. Sci. 2017, 133, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Declercq, W.; Beyaert, R.; Fiers, W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 1995, 5, 392–399. [Google Scholar] [CrossRef]
- Bazzoni, F.; Beutler, B. The tumor necrosis factor ligand and receptor families. N. Engl. J. Med. 1996, 334, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Ashkenazi, A. Tumor necrosis factor: An apoptosis JuNKie? Cell 2004, 116, 491–497. [Google Scholar] [CrossRef]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Hoffmann, A. Crosstalk via the NF-κB signaling system. Cytokine Growth Factor Rev. 2008, 19, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.A.; O’Hara, M.; Angel, P.; Chojkier, M.; Karin, M. Prolonged activation of jun and collagenase genes by tumour necrosis factor-α. Nature 1989, 337, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996, 14, 397–440. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. Cytokine expression and cell activation in inflammatory arthritis. Adv. Immunol. 1996, 63, 337–376. [Google Scholar] [PubMed]
- Keystone, E.C. Advances in targeted therapy: Safety of biological agents. Ann. Rheum. Dis. 2003, 62, ii34–ii36. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Katsikis, P.; Brennan, F.M.; Walker, J.; Bijl, H.; Ghrayeb, J.; et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor α. Arthritis Rheum. 1993, 36, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Van de Putte, L.B.; Rau, R.; Breedveld, F.C.; Kalden, J.R.; Malaise, M.G.; van Riel, P.L.; Schattenkirchner, M.; Emery, P.; Burmester, G.R.; Zeidler, H.; et al. Efficacy and safety of the fully human anti-tumour necrosis factor α monoclonal antibody adalimumab (D2E7) in DMARD refractory patients with rheumatoid arthritis: A 12 week, phase II study. Ann. Rheum. Dis. 2003, 62, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; van der Heijde, D.; de Jager, J.P.; Gough, A.; Kalden, J.; Malaise, M.; Martin Mola, E.; Pavelka, K.; Sany, J.; Settas, L.; et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: Double-blind randomised controlled trial. Lancet 2004, 363, 675–681. [Google Scholar] [CrossRef]
- Kindler, V.; Sappino, A.P.; Grau, G.E.; Piguet, P.F.; Vassalli, P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 1989, 56, 731–740. [Google Scholar] [CrossRef]
- Chen, W.; Mills, J.W.; Harmsen, A.G. Development and resolution of Pneumocystis carinii pneumonia in severe combined immunodeficient mice: A morphological study of host inflammatory responses. Int. J. Exp. Pathol. 1992, 73, 709–720. [Google Scholar] [PubMed]
- Bean, A.G.; Roach, D.R.; Briscoe, H.; France, M.P.; Korner, H.; Sedgwick, J.D.; Britton, W.J. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 1999, 162, 3504–3511. [Google Scholar] [PubMed]
- True, D.G.; Penmetcha, M.; Peckham, S.J. Disseminated cryptococcal infection in rheumatoid arthritis treated with methotrexate and infliximab. J. Rheumatol. 2002, 29, 1561–1563. [Google Scholar] [PubMed]
- van Vollenhoven, R.F.; Fleischmann, R.; Cohen, S.; Lee, E.B.; Garcia Meijide, J.A.; Wagner, S.; Forejtova, S.; Zwillich, S.H.; Gruben, D.; Koncz, T.; et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gao, J.S.; Guan, Y.; Shi, X.; Zhang, H.; Ayrapetov, M.K.; Zhang, Z.; Xu, L.; Hyun, Y.M.; Kim, M.; et al. Acetylation modulates prolactin receptor dimerization. Proc. Natl. Acad. Sci. USA 2010, 107, 19314–19319. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Kuwabara, T.; Eguchi, T.; Nakajima, K.; Kondo, M. Acetylaion regulates the MKK4-JNK pathway in T cell receptor signaling. Immunol. Lett. 2018, 194, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, D.R.; Marmorstein, R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J. 2013, 280, 5570–5581. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Itoh, G.; Kanno, S.; Uchida, K.S.; Chiba, S.; Sugino, S.; Watanabe, K.; Mizuno, K.; Yasui, A.; Hirota, T.; Tanaka, K. CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J. 2011, 30, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Akella, J.S.; Wloga, D.; Kim, J.; Starostina, N.G.; Lyons-Abbott, S.; Morrissette, N.S.; Dougan, S.T.; Kipreos, E.T.; Gaertig, J. MEC-17 is an α-tubulin acetyltransferase. Nature 2010, 467, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Schwindling, C.; Wenning, A.S.; Becherer, U.; Rettig, J.; Schwarz, E.C.; Hoth, M. T cell activation requires mitochondrial translocation to the immunological synapse. Proc. Natl. Acad. Sci. USA 2007, 104, 14418–14423. [Google Scholar] [CrossRef] [PubMed]
- Serrador, J.M.; Cabrero, J.R.; Sancho, D.; Mittelbrunn, M.; Urzainqui, A.; Sanchez-Madrid, F. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity 2004, 20, 417–428. [Google Scholar] [CrossRef]
- Hashimoto-Tane, A.; Yokosuka, T.; Sakata-Sogawa, K.; Sakuma, M.; Ishihara, C.; Tokunaga, M.; Saito, T. Dynein-driven transport of T cell receptor microclusters regulates immune synapse formation and T cell activation. Immunity 2011, 34, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuwabara, T.; Matsui, Y.; Ishikawa, F.; Kondo, M. Regulation of T-Cell Signaling by Post-Translational Modifications in Autoimmune Disease. Int. J. Mol. Sci. 2018, 19, 819. https://doi.org/10.3390/ijms19030819
Kuwabara T, Matsui Y, Ishikawa F, Kondo M. Regulation of T-Cell Signaling by Post-Translational Modifications in Autoimmune Disease. International Journal of Molecular Sciences. 2018; 19(3):819. https://doi.org/10.3390/ijms19030819
Chicago/Turabian StyleKuwabara, Taku, Yukihide Matsui, Fumio Ishikawa, and Motonari Kondo. 2018. "Regulation of T-Cell Signaling by Post-Translational Modifications in Autoimmune Disease" International Journal of Molecular Sciences 19, no. 3: 819. https://doi.org/10.3390/ijms19030819
APA StyleKuwabara, T., Matsui, Y., Ishikawa, F., & Kondo, M. (2018). Regulation of T-Cell Signaling by Post-Translational Modifications in Autoimmune Disease. International Journal of Molecular Sciences, 19(3), 819. https://doi.org/10.3390/ijms19030819