The Recombinant Inhibitor of DNA Binding Id2 Forms Multimeric Structures via the Helix-Loop-Helix Domain and the Nuclear Export Signal

 , , ,
, , ,  ,
,
Abstract
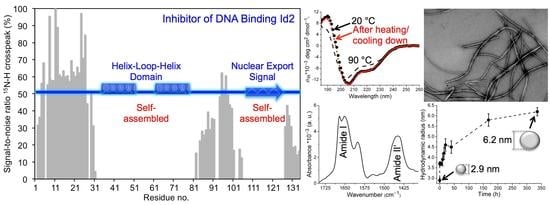
:
1. Introduction
2. Results and Discussion
2.1. Mass Spectrometry of Recombinant Id2 Protein
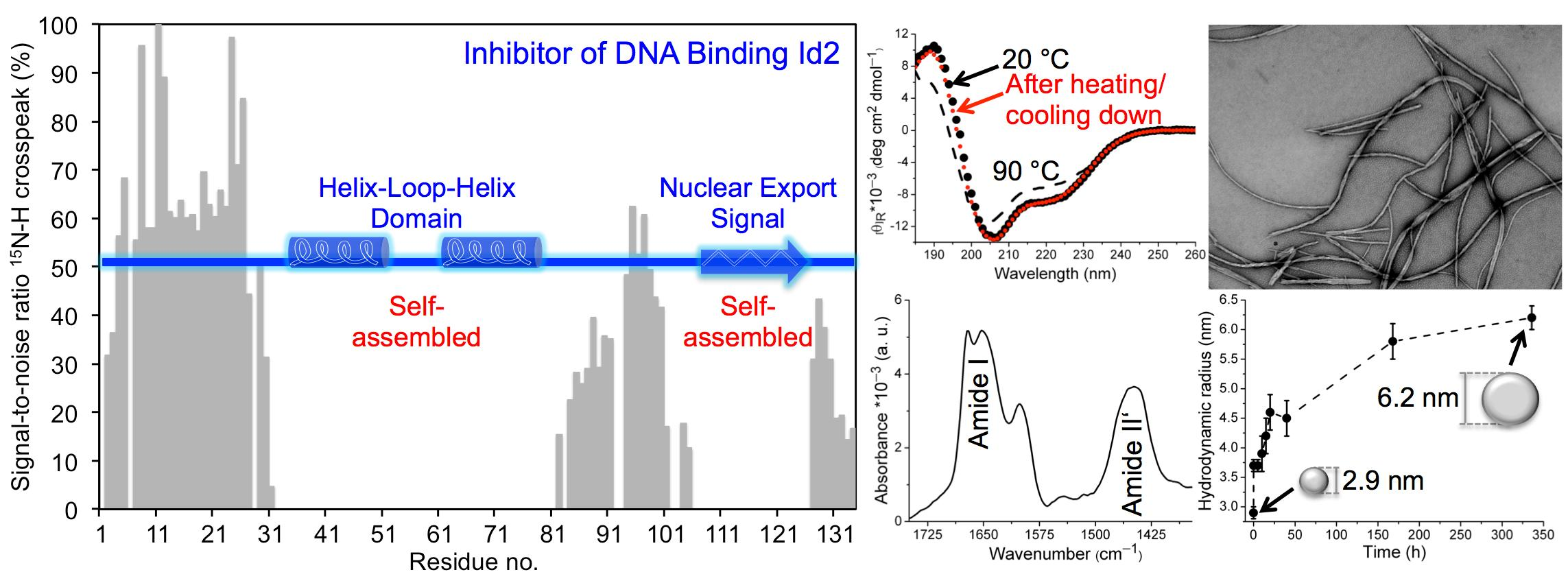
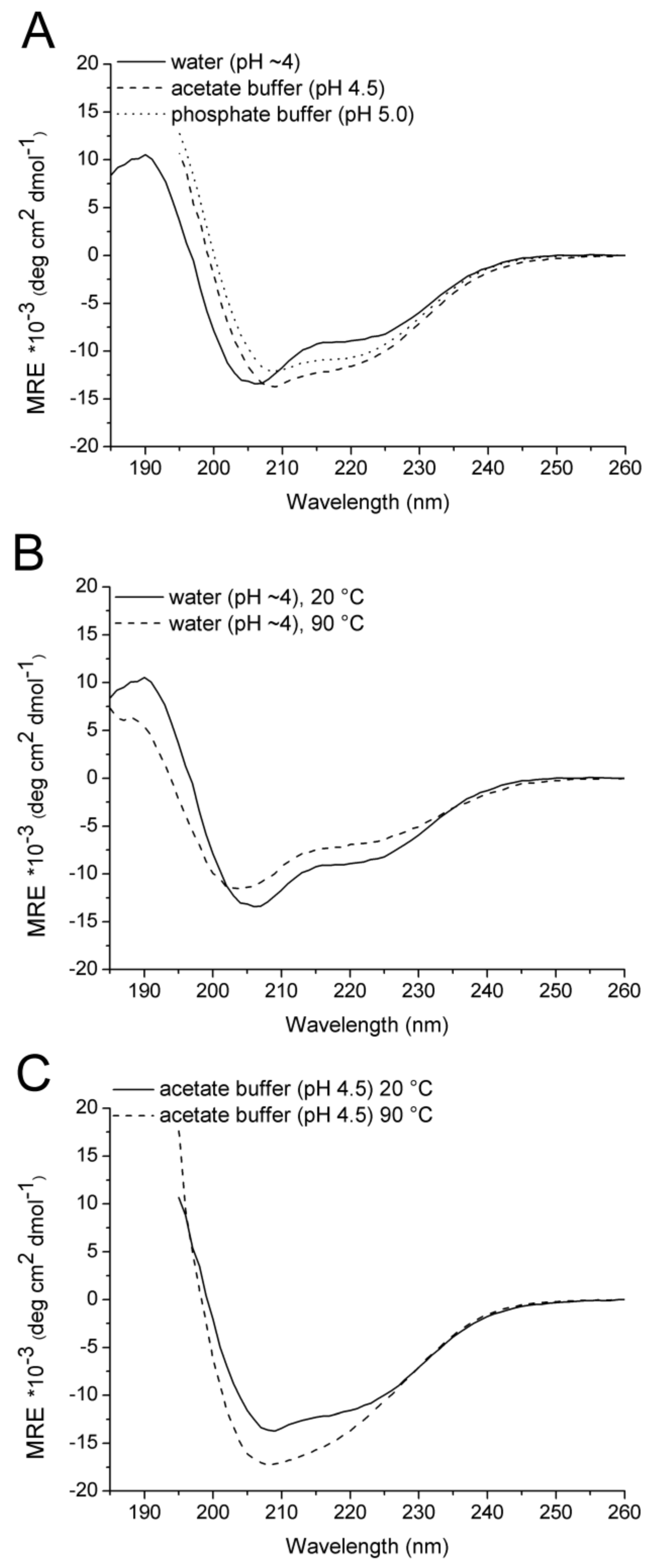
2.2. CD Spectroscopy
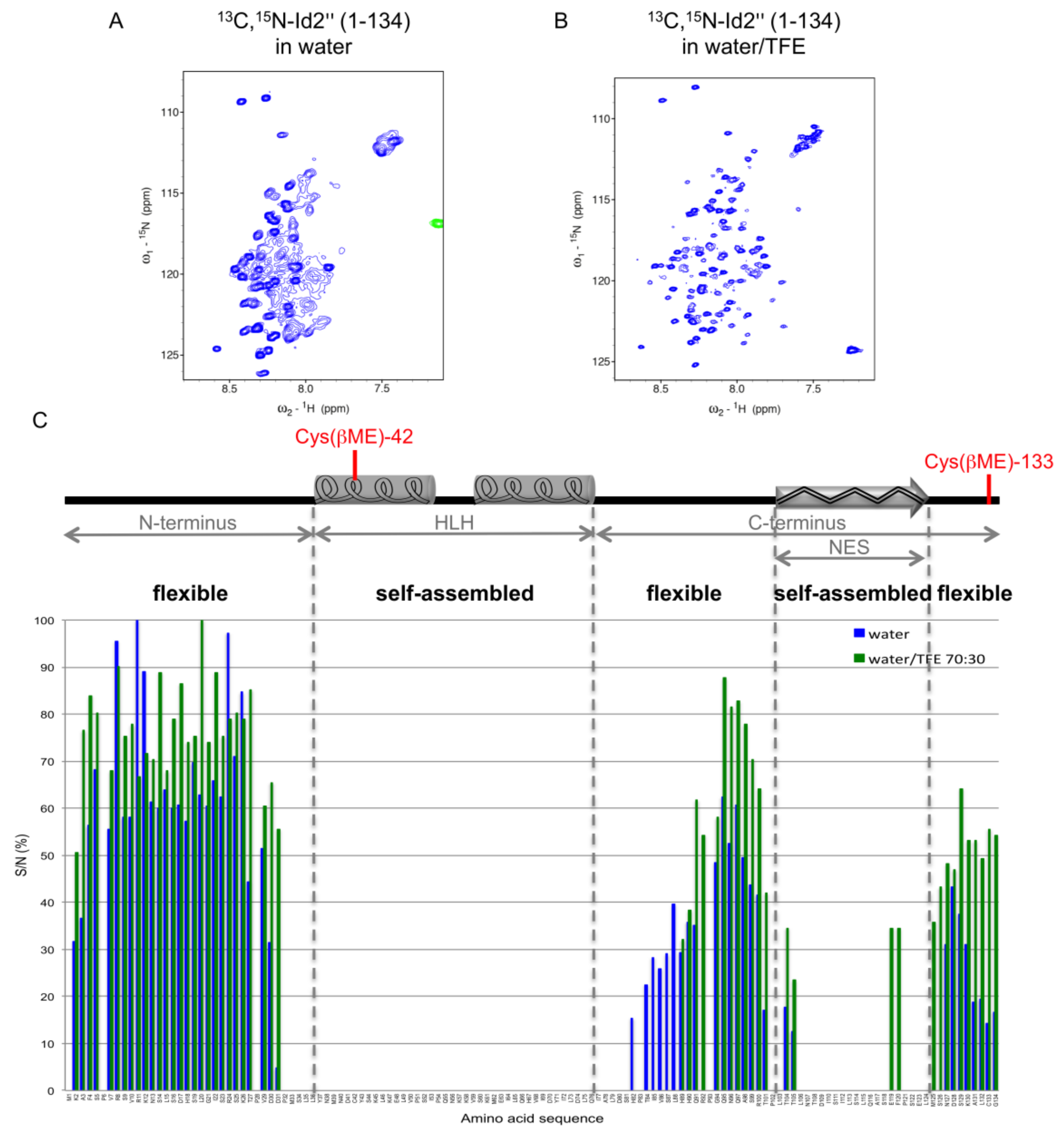
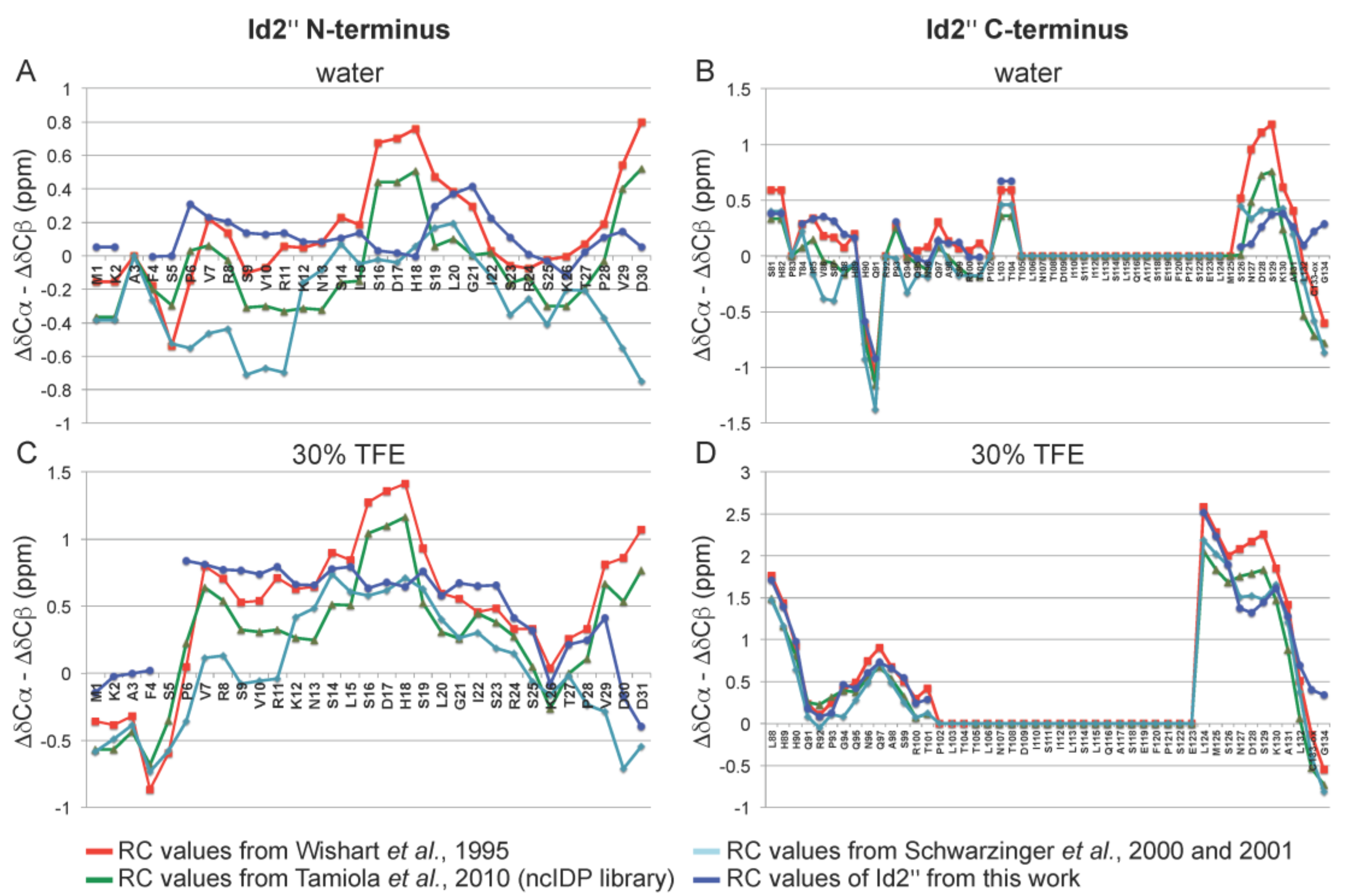
2.3. NMR Spectroscopy
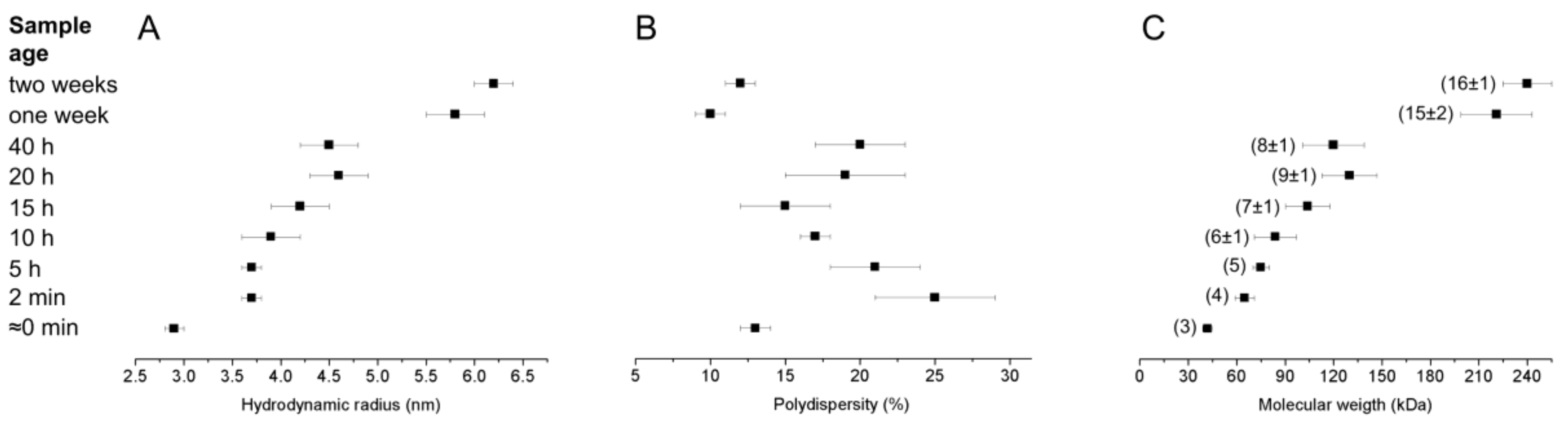
2.4. Dynamic Light Scattering
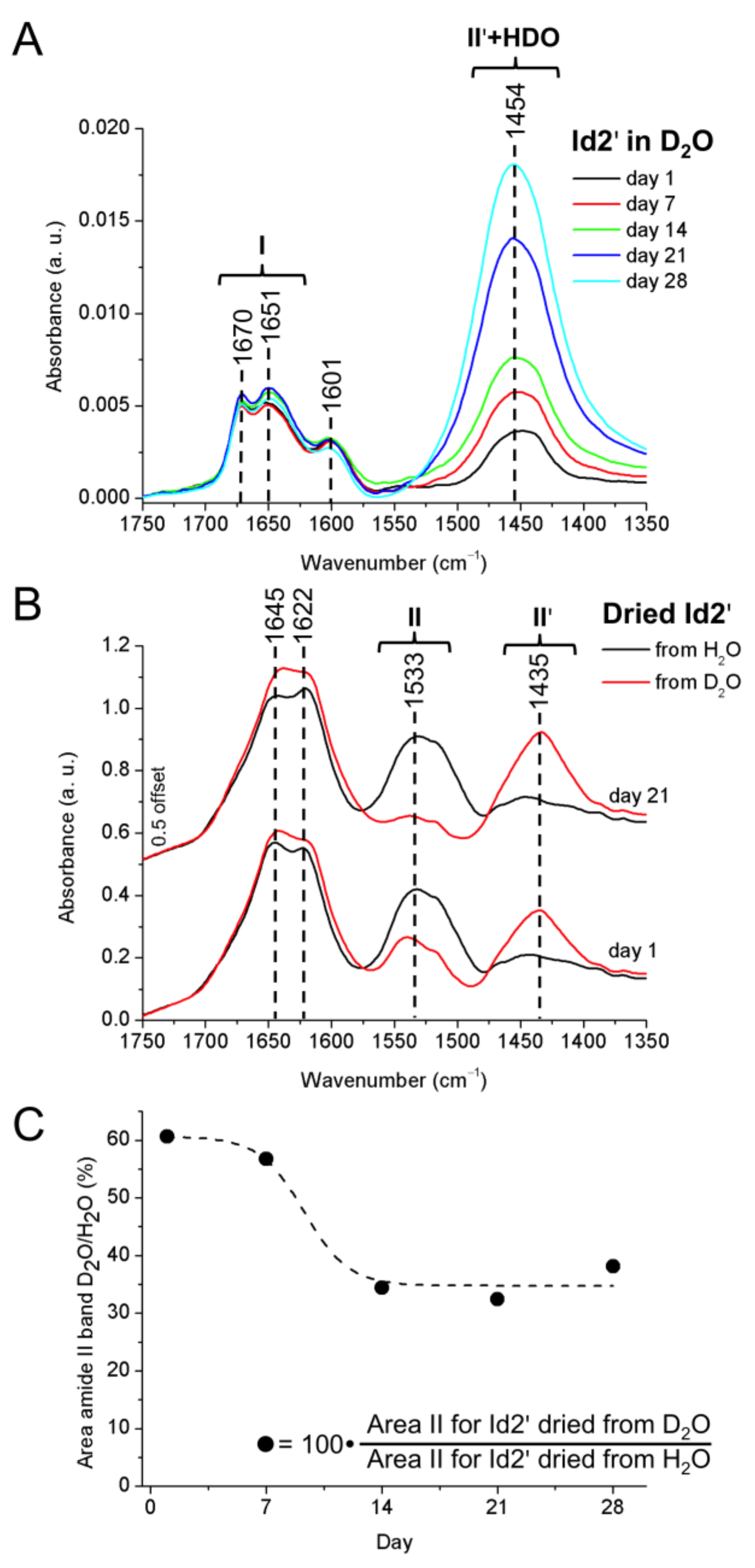
2.5. ATR-FTIR Spectroscopy
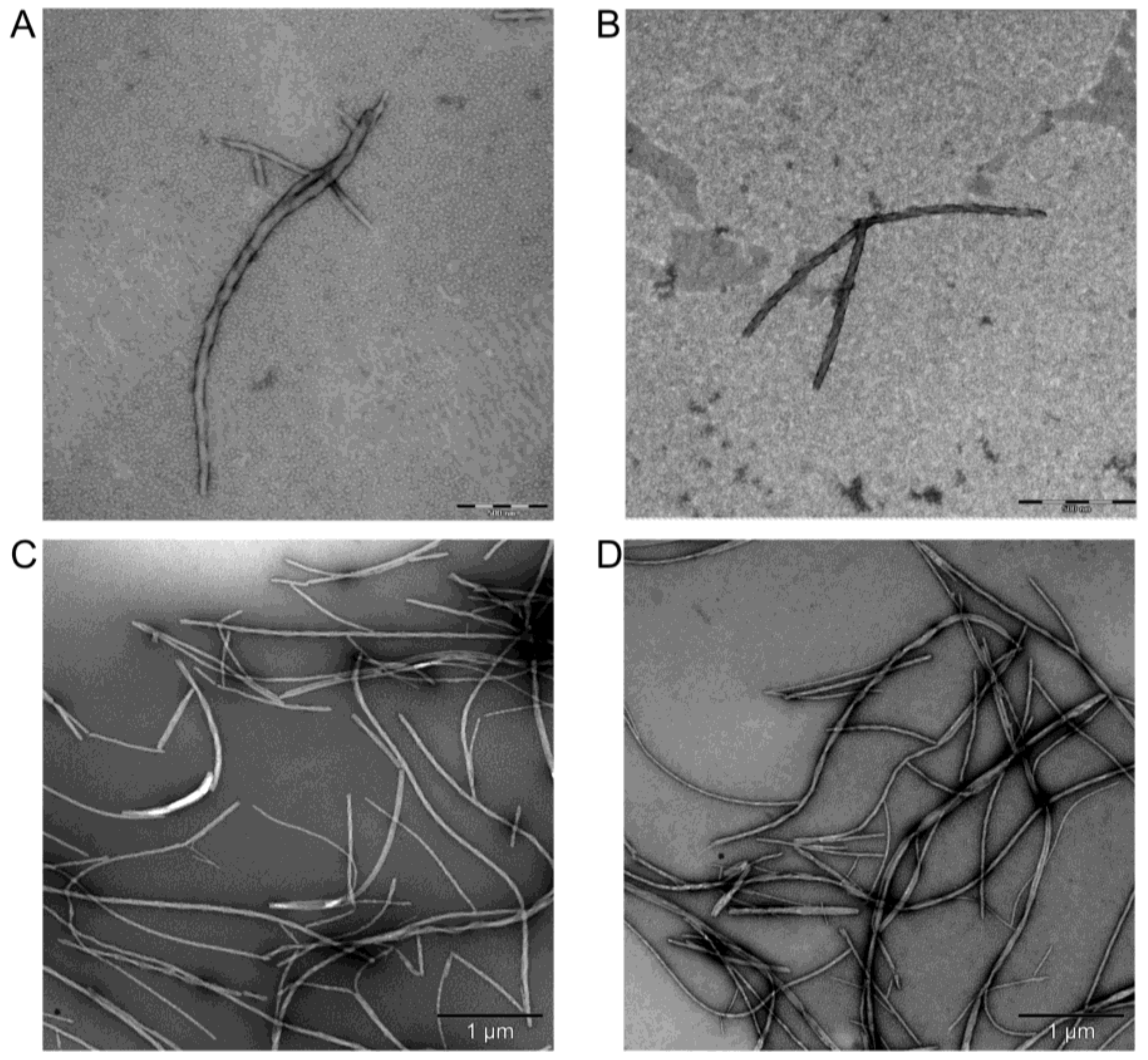
2.6. Transmission Electron Microscopy
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Construction of the pET Expression Plasmids Encoding Human Id2 1–134
4.2.2. Expression of Unlabeled and 13C,15N-Labeled Recombinant Human Id2 1–134
4.2.3. Protein Purification
4.2.4. Mass Spectrometry
4.2.5. Buffer Screening
4.2.6. CD Spectroscopy
4.2.7. NMR Spectroscopy
4.2.8. Dynamic Light Scattering
4.2.9. ATR-FTIR Spectroscopy
4.2.10. Transmission Electron Microscopy
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATR | Attenuated Total Reflection |
| CD | Circular Dichroism |
| DLS | Dynamic Light Scattering |
| HLH | Helix-Loop-Helix |
| Id2 | Inhibitor of DNA Binding and Cell Differentiation 2 |
| IDP | Intrinsically Disordered Protein |
| MCT | Mercury Cadmium Telluride |
| βME | β-Mercaptoethanol |
| TEM | Transmission Electron Microscopy |
| TFA | Trifluoroacetic Acid |
References
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 2000, 20, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Fass, D.; Arnaud, M.; Harrison, S.C. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev. 1994, 8, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Rould, M.A.; Weintraub, H.; Pabo, C.O. Crystal structure of MyoD bHLH domain-DNA complex: Perspectives on DNA recognition and implications for transcriptional activation. Cell 1994, 77, 451–459. [Google Scholar] [CrossRef]
- Ferre-D’Amare, A.R.; Prendergast, G.C.; Ziff, E.B.; Burley, S.K. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 1993, 363, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Anand, G.; Yin, X.; Shahidi, A.K.; Grove, L.; Prochownik, E.V. Novel regulation of the helix-loop-helix protein Id1 by S5a, a subunit of the 26 S proteasome. J. Biol. Chem. 1997, 272, 19140–19151. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, A.; Garg, P.; Lasorella, A.; Hsu, J.; Israel, M.A. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994, 8, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Iavarone, A.; Israel, M.A. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol. Cell. Biol. 1996, 16, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Yates, P.R.; Atherton, G.T.; Deed, R.W.; Norton, J.D.; Sharrocks, A.D. Id helix-loop-helix proteins inhibit nucleoprotein complex formation by the TCF ETS-domain transcription factors. EMBO J. 1999, 18, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Stegmuller, J.; Guardavaccaro, D.; Liu, G.; Carro, M.S.; Rothschild, G.; de la Torre-Ubieta, L.; Pagano, M.; Bonni, A.; Iavarone, A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 2006, 442, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An Id2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, W.; Warburton, D. A cysteine residue in the helix-loop-helix domain of Id2 is critical for homodimerization and function. Biochem. Biophys. Res. Commun. 2000, 273, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Wibley, J.; Deed, R.; Jasiok, M.; Douglas, K.; Norton, J. A homology model of the Id-3 helix-loop-helix domain as a basis for structure-function predictions. Biochim. Biophys. Acta 1996, 1294, 138–146. [Google Scholar] [CrossRef]
- Loveys, D.A.; Streiff, M.B.; Kato, G.J. E2A basic-helix-loop-helix transcription factors are negatively regulated by serum growth factors and by the Id3 protein. Nucleic Acids Res. 1996, 24, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Chinaranagari, S.; Chaudhary, J. Inhibitor of Differentiation 4 (ID4) Acts as an Inhibitor of ID-1, -2 and -3 and Promotes basic Helix Loop Helix (bHLH) E47 DNA Binding and Transcriptional Activity. Biochimie 2015, 112, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J. Expanding the proteome: Disordered and alternatively folded proteins. Q. Rev. Biophys. 2011, 44, 467–518. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P.; Simon, I.; Uversky, V.N.; Hansen, J.C.; Asturias, F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008, 4, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Svobodova, J.; Cabrele, C. Stepwise solid-phase synthesis and spontaneous homodimerization of the helix-loop-helix protein Id3. ChemBioChem 2006, 7, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, M.; Yoshiya, T.; Kiso, Y.; Cabrele, C. Synthesis and conformation of an analog of the helix-loop-helix domain of the Id1 protein containing the O-acyl iso-prolyl-seryl switch motif. J. Pept. Sci. 2010, 16, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Cabrele, C. Synthesis and conformational analysis of Id2 protein fragments: Impact of chain length and point mutations on the structural HLH motif. J. Pept. Sci. 2006, 12, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Fairman, R.; Beran-Steed, R.K.; Anthony-Cahill, S.J.; Lear, J.D.; Stafford, W.F., 3rd; DeGrado, W.F.; Benfield, P.A.; Brenner, S.L. Multiple oligomeric states regulate the DNA binding of helix-loop-helix peptides. Proc. Natl. Acad. Sci. USA 1993, 90, 10429–10433. [Google Scholar] [CrossRef] [PubMed]
- Kiewitz, S.D.; Cabrele, C. Synthesis and conformational properties of protein fragments based on the Id family of DNA-binding and cell-differentiation inhibitors. Biopolymers 2005, 80, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Kiewitz, S.D.; Kakizawa, T.; Kiso, Y.; Cabrele, C. Switching from the unfolded to the folded state of the helix-loop-helix domain of the Id proteins based on the O-acyl isopeptide method. J. Pept. Sci. 2008, 14, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.V.; Jiang, S.; Palasingam, P.; Kolatkar, P.R. A divalent ion is crucial in the structure and dominant-negative function of ID proteins, a class of helix-loop-helix transcription regulators. PLoS ONE 2012, 7, e48591. [Google Scholar] [CrossRef] [PubMed]
- Eletsky, A.; Wang, D.; Kohan, E.; Janjua, H.; Acton, T.B.; Xiao, R.; Everett, J.K.; Montelione, G.T.; Szyperski, T. Solution NMR Structure of the Helix-Loop-Helix Domain of Human ID3 Protein, Northeast Structural Genomics Consortium Target HR3111A. Available online: http://www.ebi.ac.uk/pdbe/entry/pdb/2LFH (accessed on 27 July 2011).
- Beisswenger, M.; Cabrele, C. Self-recognition behavior of a helix-loop-helix domain by a fragment scan. Biochim. Biophys. Acta 2014, 1844, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W.; Glockner, J. Estimation of Globular Protein Secondary Structure from Circular-Dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Van Stokkum, I.H.; Spoelder, H.J.; Bloemendal, M.; van Grondelle, R.; Groen, F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990, 191, 110–118. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Roccatano, D.; Colombo, G.; Fioroni, M.; Mark, A.E. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study. Proc. Natl. Acad. Sci. USA 2002, 99, 12179–12184. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Schmid, F.X. Stabilization of a protein by guanidinium chloride. Biochemistry 1993, 32, 7994–7998. [Google Scholar] [CrossRef] [PubMed]
- Zarrine-Afsar, A.; Mittermaier, A.; Kay, L.E.; Davidson, A.R. Protein stabilization by specific binding of guanidinium to a functional arginine-binding surface on an SH3 domain. Protein Sci. 2006, 15, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Kurooka, H.; Yokota, Y. Nucleo-cytoplasmic shuttling of Id2, a negative regulator of basic helix-loop-helix transcription factors. J. Biol. Chem. 2005, 280, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Schroeder, J.; Cabrele, C. A short Id2 protein fragment containing the nuclear export signal forms amyloid-like fibrils. Biochem. Biophys. Res. Commun. 2006, 346, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Weisemann, R.; Ruterjans, H.; Bermel, W. 3D triple-resonance NMR techniques for the sequential assignment of NH and 15N resonances in 15N- and 13C-labelled proteins. J. Biomol. NMR 1993, 3, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Smalla, M.; Schmieder, P.; Oschkinat, H. MUSIC in triple-resonance experiments: Amino acid type-selective (1)H-(15)N correlations. J. Magn. Reson. 1999, 141, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Schwarzinger, S.; Kroon, G.J.; Foss, T.R.; Chung, J.; Wright, P.E.; Dyson, H.J. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001, 123, 2970–2978. [Google Scholar] [CrossRef] [PubMed]
- Schwarzinger, S.; Kroon, G.J.; Foss, T.R.; Wright, P.E.; Dyson, H.J. Random coil chemical shifts in acidic 8 M urea: Implementation of random coil shift data in NMRView. J. Biomol. NMR 2000, 18, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Holm, A.; Hodges, R.S.; Sykes, B.D. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995, 5, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Tamiola, K.; Acar, B.; Mulder, F.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 2010, 132, 18000–18003. [Google Scholar] [CrossRef] [PubMed]
- Roschger, C.; Cabrele, C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun. Signal. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Metzler, W.J.; Constantine, K.L.; Friedrichs, M.S.; Bell, A.J.; Ernst, E.G.; Lavoie, T.B.; Mueller, L. Characterization of the three-dimensional solution structure of human profilin: 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry 1993, 32, 13818–13829. [Google Scholar] [CrossRef] [PubMed]
- Khurshid, S.; Saridakis, E.; Govada, L.; Chayen, N.E. Porous nucleating agents for protein crystallization. Nat. Protoc. 2014, 9, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.K.; Swain, T.; Chandra, N.; Swaminathan, R. On the characterization of intermediates in the isodesmic aggregation pathway of hen lysozyme at alkaline pH. PLoS ONE 2014, 9, e87256. [Google Scholar] [CrossRef] [PubMed]
- Laue, T.M.; Starovasnik, M.A.; Weintraub, H.; Sun, X.H.; Snider, L.; Klevit, R.E. MyoD forms micelles which can dissociate to form heterodimers with E47: Implications of micellization on function. Proc. Natl. Acad. Sci. USA 1995, 92, 11824–11828. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient protein-protein interactions: Structural, functional, and network properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Arvan, P.; Zhang, B.Y.; Feng, L.; Liu, M.; Kuliawat, R. Lumenal protein multimerization in the distal secretory pathway/secretory granules. Curr. Opin. Cell Biol. 2002, 14, 448–453. [Google Scholar] [CrossRef]
- Dannies, P.S. Concentrating hormones into secretory granules: Layers of control. Mol. Cell. Endocrinol. 2001, 177, 87–93. [Google Scholar] [CrossRef]
- Keeler, C.; Hodsdon, M.E.; Dannies, P.S. Is there structural specificity in the reversible protein aggregates that are stored in secretory granules? J. Mol. Neurosci. 2004, 22, 43–49. [Google Scholar] [CrossRef]
- Tooze, S.A. Biogenesis of secretory granules in the trans-Golgi network of neuroendocrine and endocrine cells. Biochim. Biophys. Acta 1998, 1404, 231–244. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Barth, A.; Zscherp, C. What vibrations tell us aboutproteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Molecular Cloning, 4 ed.; Cold Spring Harbour Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Lepre, C.A.; Moore, J.M. Microdrop screening: A rapid method to optimize solvent conditions for NMR spectroscopy of proteins. J. Biomol. NMR 1998, 12, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Mach, H.; Middaugh, C.R.; Lewis, R.V. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal. Biochem. 1992, 200, 74–80. [Google Scholar] [CrossRef]
- Sattler, M.; Schleucher, J.; Griesinger, C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsedfield gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar] [CrossRef]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wuthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J. Biomol. NMR 1998, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Márquez, A.; Kocsis, K.; Zickler, G.; Bourret, G.R.; Feinle, A.; Hüsing, N.; Himly, M.; Duschl, A.; Berger, T.; Diwald, O. Enzyme adsorption-induced activity changes: A quantitative study on TiO2 model agglomerates. J. Nanobiotechnol. 2017, 15, 55. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Temperature (°C) | α (%) | β (%) | Turn (%) | Disordered (%) |
|---|---|---|---|---|---|
| Water | 20 | 28 | 12 | 20 | 40 |
| Water/50% TFE | 20 | 32 | 16 | 20 | 33 |
| Water | 90 | 19 | 14 | 15 | 52 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roschger, C.; Schubert, M.; Regl, C.; Andosch, A.; Marquez, A.; Berger, T.; Huber, C.G.; Lütz-Meindl, U.; Cabrele, C. The Recombinant Inhibitor of DNA Binding Id2 Forms Multimeric Structures via the Helix-Loop-Helix Domain and the Nuclear Export Signal. Int. J. Mol. Sci. 2018, 19, 1105. https://doi.org/10.3390/ijms19041105
Roschger C, Schubert M, Regl C, Andosch A, Marquez A, Berger T, Huber CG, Lütz-Meindl U, Cabrele C. The Recombinant Inhibitor of DNA Binding Id2 Forms Multimeric Structures via the Helix-Loop-Helix Domain and the Nuclear Export Signal. International Journal of Molecular Sciences. 2018; 19(4):1105. https://doi.org/10.3390/ijms19041105
Chicago/Turabian StyleRoschger, Cornelia, Mario Schubert, Christof Regl, Ancuela Andosch, Augusto Marquez, Thomas Berger, Christian G. Huber, Ursula Lütz-Meindl, and Chiara Cabrele. 2018. "The Recombinant Inhibitor of DNA Binding Id2 Forms Multimeric Structures via the Helix-Loop-Helix Domain and the Nuclear Export Signal" International Journal of Molecular Sciences 19, no. 4: 1105. https://doi.org/10.3390/ijms19041105
APA StyleRoschger, C., Schubert, M., Regl, C., Andosch, A., Marquez, A., Berger, T., Huber, C. G., Lütz-Meindl, U., & Cabrele, C. (2018). The Recombinant Inhibitor of DNA Binding Id2 Forms Multimeric Structures via the Helix-Loop-Helix Domain and the Nuclear Export Signal. International Journal of Molecular Sciences, 19(4), 1105. https://doi.org/10.3390/ijms19041105