Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment

Abstract
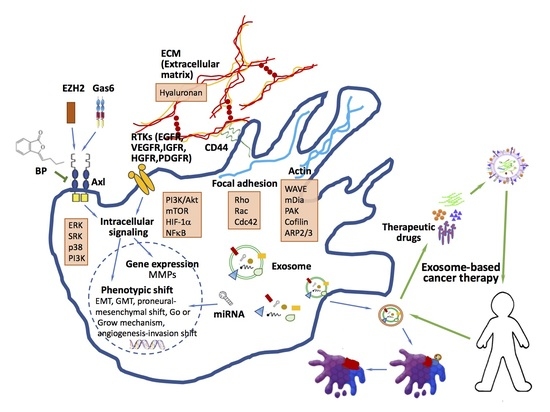
:
1. Introduction
2. Cancer Stem Cell Contribution to Glioblastoma Invasiveness
3. Phenotypic Shift in GBM
3.1. Epithelial-Mesenchymal Transition (EMT) and Metastasis
3.2. Proneural-Mesenchymal Shift
3.3. Migration/Proliferation Dichotomy (Go or Grow Mechanism)
3.4. Angiogenesis-Invasion Shift
3.5. Glial-Mesenchymal Transition (GMT)
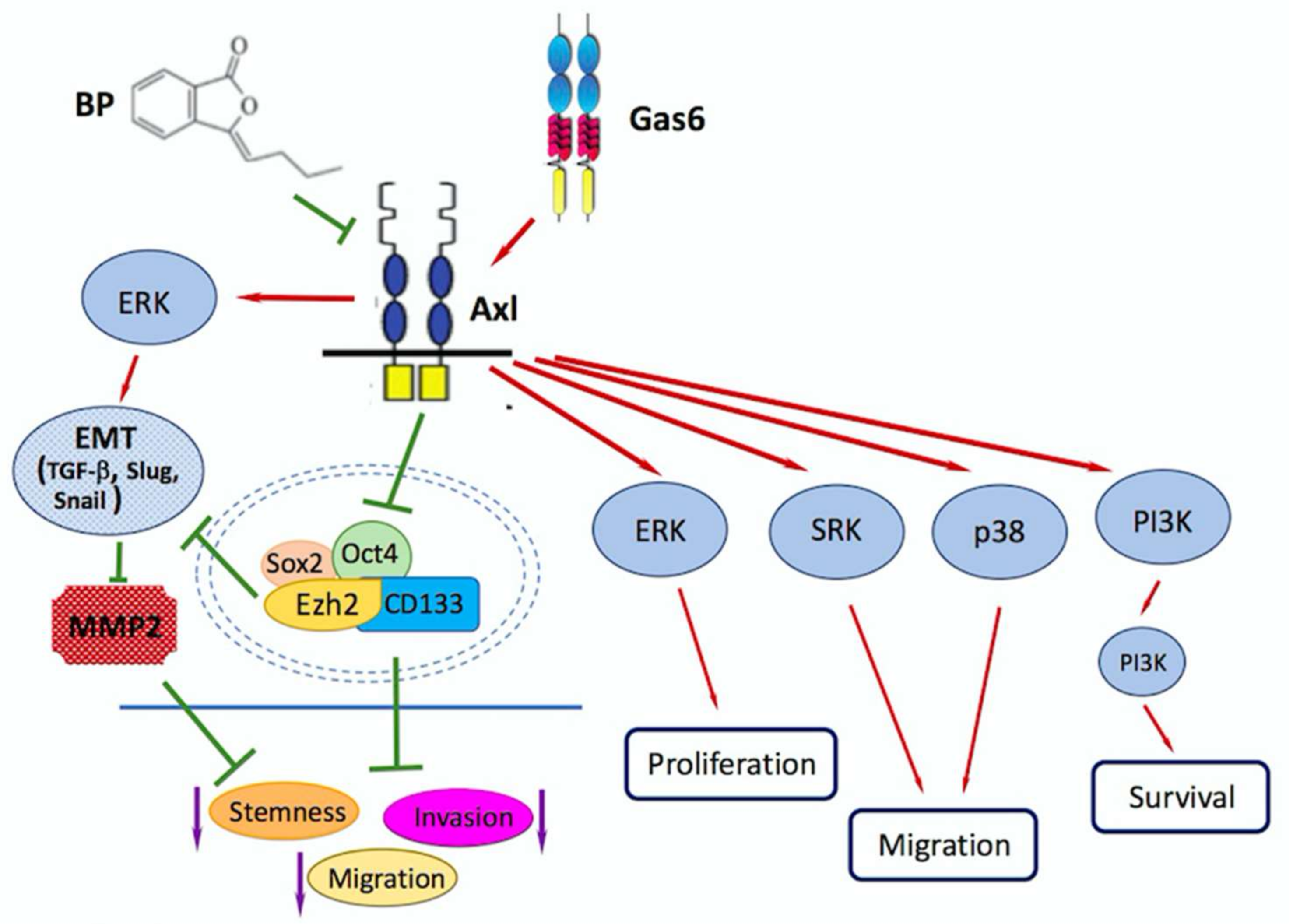
4. Role of EZH2/AXL/TGF-β axis in Glioblastoma Metastasis/Invasion
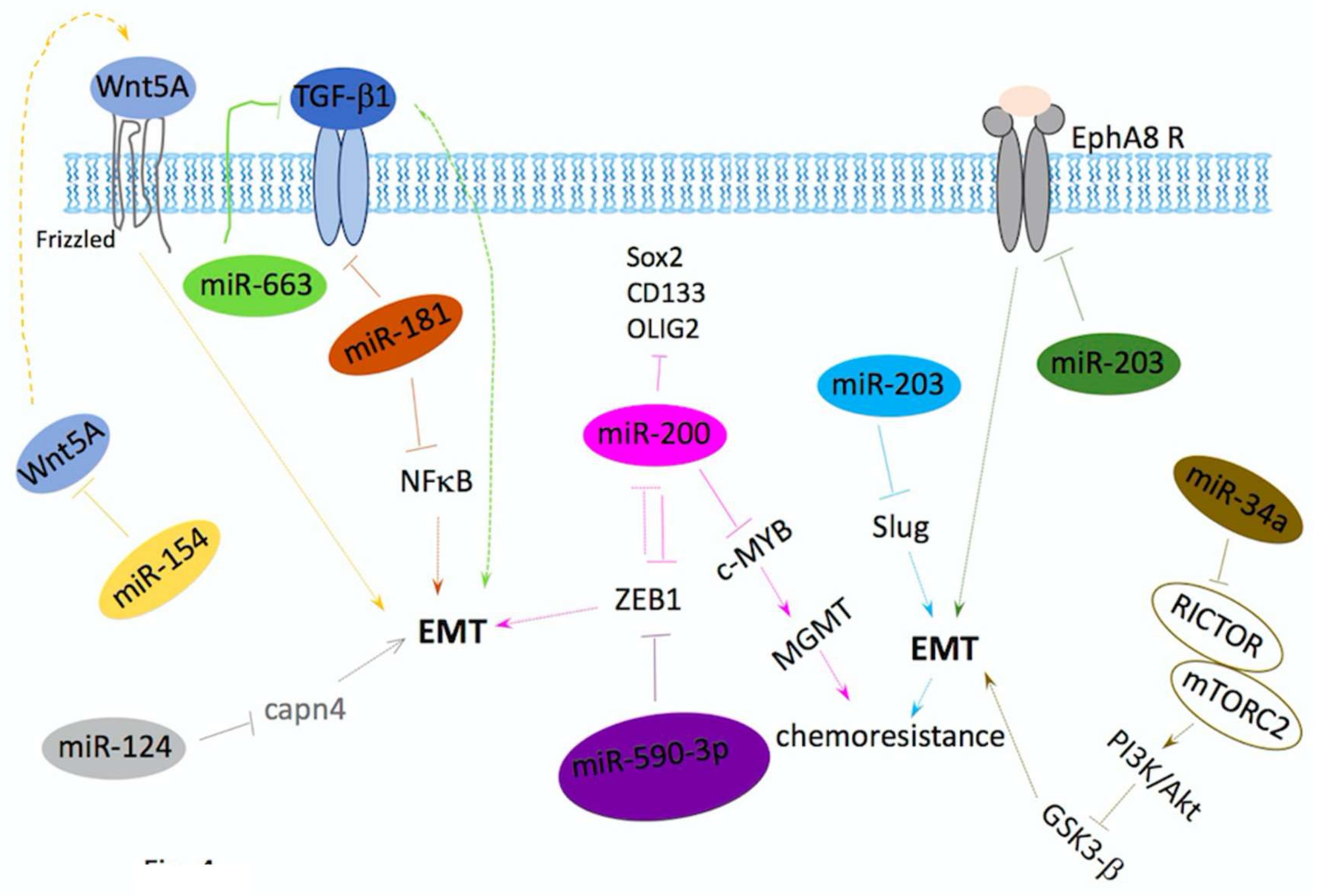
5. miRNAs/Drugs in Inhibition of Invasion and Metastasis
miRNAs Regulate EMT in GBM
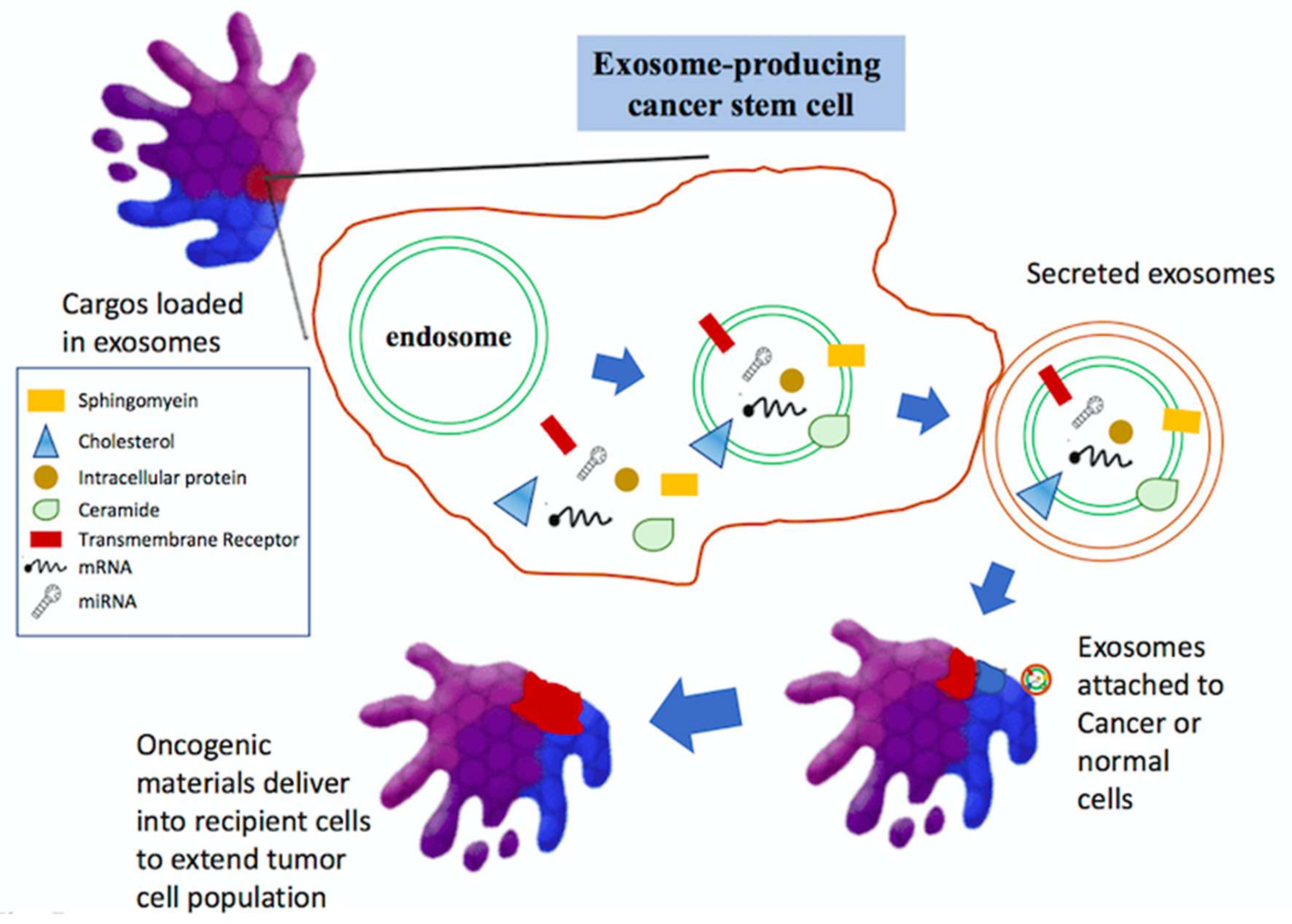
6. Exosomes in Glioma Progression and Therapeutic Potentials
7. Role of Exosome in Tumors
7.1. Tumorigenesis
7.2. Tumor Metastasis
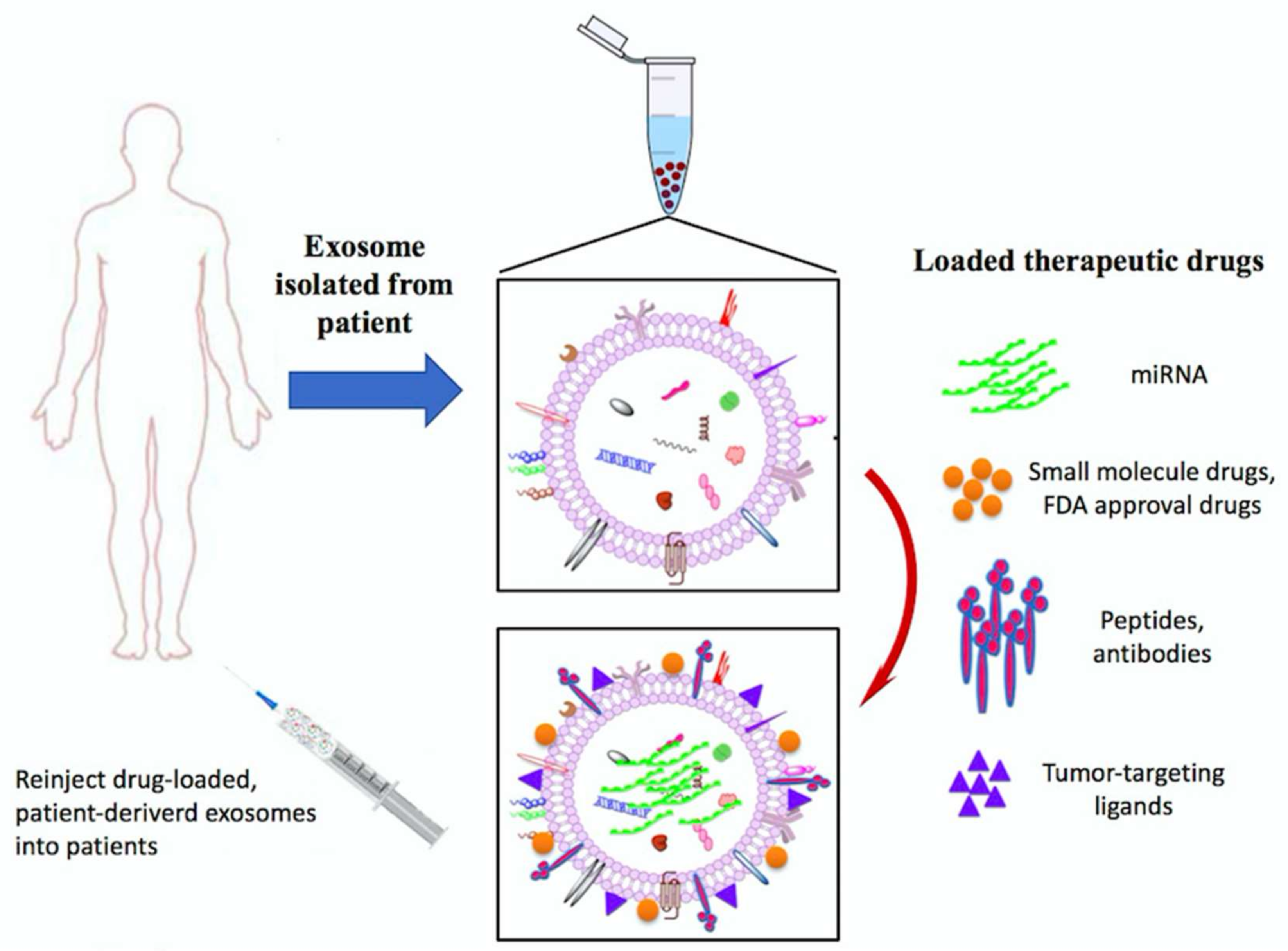
7.3. Exosomes Application as Anti-Cancer Drug Delivery Vehicles
8. Summary and Future Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Behin, A.; Hoang-Xuan, K.; Carpentier, A.F.; Delattre, J.Y. Primary brain tumours in adults. Lancet 2003, 361, 323–331. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Vengoechea, J.; Zheng, S.; Sloan, A.E.; Chen, Y.; Brat, D.J.; O’Neill, B.P.; de Groot, J.; Yust-Katz, S.; Yung, W.K.; et al. Molecular subtypes of glioblastoma are relevant to lower grade glioma. PLoS ONE 2014, 9, e91216. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research, N., Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, L.; Hong, C.S.; Yang, C.; Zhuang, Z.; Heiss, J.D. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int. J. Med. Sci. 2015, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; van Noorden, C.J.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta 2014, 1846, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Pirozzi, C.J.; Lopez, G.Y.; Yan, H. Isocitrate dehydrogenase mutations in gliomas: Mechanisms, biomarkers and therapeutic target. Curr. Opin. Neurol. 2011, 24, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Margittai, E.; Banhegyi, G. Isocitrate dehydrogenase: A NADPH-generating enzyme in the lumen of the endoplasmic reticulum. Arch. Biochem. Biophys. 2008, 471, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Turkalp, Z.; Karamchandani, J.; Das, S. IDH mutation in glioma: New insights and promises for the future. JAMA Neurol. 2014, 71, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ling, Z.Q. Role of isocitrate dehydrogenase 1/2 (IDH 1/2) gene mutations in human tumors. Histol. Histopathol. 2015, 30, 1155–1160. [Google Scholar] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Mehta, M.P. Role of isocitrate dehydrogenase in glioma. Expert Rev. Neurother. 2011, 11, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Redhu, R.; Nadkarni, T.; Goel, A. Supratentorial glioblastoma multiforme with spinal metastases. J. Craniovertebr. Junction Spine 2010, 1, 126–129. [Google Scholar] [PubMed]
- Vertosick, F.T., Jr.; Selker, R.G. Brain stem and spinal metastases of supratentorial glioblastoma multiforme: A clinical series. Neurosurgery 1990, 27, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.D.; Rapp, M.; Schneiderhan, T.; Sabel, M.; Hayman, A.; Scherer, A.; Kropil, P.; Budach, W.; Gerber, P.; Kretschmar, U.; et al. Glioblastoma multiforme metastasis outside the CNS: Three case reports and possible mechanisms of escape. J. Clin. Oncol. 2014, 32, e80–e84. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P. Extra-neural metastases of malignant gliomas: Myth or reality? Cancers (Basel) 2011, 3, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Lun, M.; Lok, E.; Gautam, S.; Wu, E.; Wong, E.T. The natural history of extracranial metastasis from glioblastoma multiforme. J. Neurooncol. 2011, 105, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Keene, D.; Ventureyra, E.; Matzinger, M.A.; Jimenez, C.; Wang, H.S.; Grimard, L. Bone marrow metastasis in astrocytic gliomata. J. Neurooncol. 1998, 37, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Fraser, J.R. Hyaluronan. FASEB J. 1992, 6, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Delpech, B.; Maingonnat, C.; Girard, N.; Chauzy, C.; Maunoury, R.; Olivier, A.; Tayot, J.; Creissard, P. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumour stroma. Eur. J. Cancer 1993, 29A, 1012–1017. [Google Scholar] [CrossRef]
- Wiranowska, M.; Ladd, S.; Moscinski, L.C.; Hill, B.; Haller, E.; Mikecz, K.; Plaas, A. Modulation of hyaluronan production by CD44 positive glioma cells. Int. J. Cancer 2010, 127, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Wiranowska, M.; Ladd, S.; Smith, S.R.; Gottschall, P.E. CD44 adhesion molecule and neuro-glial proteoglycan NG2 as invasive markers of glioma. Brain Cell Biol. 2006, 35, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Ariza, A.; Lopez, D.; Mate, J.L.; Isamat, M.; Musulen, E.; Pujol, M.; Ley, A.; Navas-Palacios, J.J. Role of CD44 in the invasiveness of glioblastoma multiforme and the noninvasiveness of meningioma: An immunohistochemistry study. Hum. Pathol. 1995, 26, 1144–1147. [Google Scholar] [CrossRef]
- Bourguignon, L.Y. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 2008, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Zhu, H.; Shao, L.; Chen, Y.W. CD44 interaction with tiam1 promotes Rac1 signaling and hyaluronic acid-mediated breast tumor cell migration. J. Biol. Chem. 2000, 275, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Gibellini, F.; Njuguna, N.; Hazan-Halevy, I.; Farooqui, M.; Bern, S.; Keyvanfar, K.; Lee, E.; Wilson, W.; Wiestner, A. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk. Lymphoma 2011, 52, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.J.; Brangwynne, C.P.; Kasza, K.E.; Filippidi, E.; Gordon, V.D.; Deisboeck, T.S.; Weitz, D.A. Glioma expansion in collagen I matrices: Analyzing collagen concentration-dependent growth and motility patterns. Biophys. J. 2005, 89, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Kawataki, T.; Yamane, T.; Naganuma, H.; Rousselle, P.; Anduren, I.; Tryggvason, K.; Patarroyo, M. Laminin isoforms and their integrin receptors in glioma cell migration and invasiveness: Evidence for a role of α5-laminin(s) and α3β1 integrin. Exp. Cell Res. 2007, 313, 3819–3831. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.R.; Huang, P.; et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Hiraga, S.; Izumoto, S.; Matsumura, H.; Kanemura, Y.; Arita, N.; Hayakawa, T. Role of fibronectin-stimulated tumor cell migration in glioma invasion in vivo: Clinical significance of fibronectin and fibronectin receptor expressed in human glioma tissues. Clin. Exp. Metastasis 1998, 16, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, S.; Haase, M.; Cordes, N. Signalling via integrins: Implications for cell survival and anticancer strategies. Biochim. Biophys. Acta 2007, 1775, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Aoudjit, F.; Vuori, K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 2001, 20, 4995–5004. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Madani, I.; De Neve, W.; Mareel, M. Does ionizing radiation stimulate cancer invasion and metastasis? Bull. Cancer 2008, 95, 292–300. [Google Scholar] [PubMed]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, M.; Su, T.T. Radiation responses and resistance. Int. Rev. Cell Mol. Biol. 2012, 299, 235–253. [Google Scholar] [PubMed]
- Rycaj, K.; Tang, D.G. Cancer stem cells and radioresistance. Int. J. Radiat. Biol. 2014, 90, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Rojas-Sutterlin, S.; Laflamme, C.; Lachambre, M.P.; Rolland, Y.; Sartelet, H.; Beliveau, R. Tumor environment dictates medulloblastoma cancer stem cell expression and invasive phenotype. Mol. Cancer Res. 2008, 6, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Lachambre, M.P.; Plouffe, K.; Sartelet, H.; Beliveau, R. Modulation of invasive properties of CD133+ glioblastoma stem cells: A role for MT1-MMP in bioactive lysophospholipid signaling. Mol. Carcinog. 2009, 48, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Takahashi, H.; Harada, H.; Kohno, S.; Ohue, S.; Kobayashi, K.; Yano, H.; Tanaka, J.; Ohnishi, T. Cancer stem-like cells of glioblastoma characteristically express MMP-13 and display highly invasive activity. Int. J. Oncol. 2010, 37, 1121–1131. [Google Scholar] [PubMed]
- Ortensi, B.; Osti, D.; Pellegatta, S.; Pisati, F.; Brescia, P.; Fornasari, L.; Levi, D.; Gaetani, P.; Colombo, P.; Ferri, A.; et al. Rai is a new regulator of neural progenitor migration and glioblastoma invasion. Stem Cells 2012, 30, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Tamase, A.; Muraguchi, T.; Naka, K.; Tanaka, S.; Kinoshita, M.; Hoshii, T.; Ohmura, M.; Shugo, H.; Ooshio, T.; Nakada, M.; et al. Identification of tumor-initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc. Natl. Acad. Sci. USA 2009, 106, 17163–17168. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Diez-Valle, R.; Manterola, L.; Rubio, A.; Liu, D.; Cortes-Santiago, N.; Urquiza, L.; Jauregi, P.; Lopez de Munain, A.; Sampron, N.; et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE 2011, 6, e26740. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Temme, A.; Senner, V.; Ebner, R.; Schwind, S.; Stevanovic, S.; Wehner, R.; Schackert, G.; Schackert, H.K.; Fussel, M.; et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br. J. Cancer 2007, 96, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Favaro, R.; Appolloni, I.; Pellegatta, S.; Sanga, A.B.; Pagella, P.; Gambini, E.; Pisati, F.; Ottolenghi, S.; Foti, M.; Finocchiaro, G.; et al. SOX2 is required to maintain cancer stem cells in a mouse model of high-grade oligodendroglioma. Cancer Res. 2014, 74, 1833–1844. [Google Scholar] [CrossRef] [PubMed]
- Rape, A.; Ananthanarayanan, B.; Kumar, S. Engineering strategies to mimic the glioblastoma microenvironment. Adv. Drug Deliv. Rev. 2014, 79–80, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Otani, Y.; Kurozumi, K.; Date, I. Phenotypic Transition as a Survival Strategy of Glioma. Neurol. Medico Chir. (Tokyo) 2016, 56, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014, 16, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Nevo, I.; Woolard, K.; Cam, M.; Li, A.; Webster, J.D.; Kotliarov, Y.; Kim, H.S.; Ahn, S.; Walling, J.; Kotliarova, S.; et al. Identification of molecular pathways facilitating glioma cell invasion in situ. PLoS ONE 2014, 9, e111783. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Boutet, A.; Esteban, M.A.; Maxwell, P.H.; Nieto, M.A. Reactivation of Snail genes in renal fibrosis and carcinomas: A process of reversed embryogenesis? Cell Cycle 2007, 6, 638–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A.; et al. TWIST is expressed in human gliomas and promotes invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Lawton, C.D.; Nagasawa, D.T.; Yang, I.; Fessler, R.G.; Smith, Z.A. Leptomeningeal spinal metastases from glioblastoma multiforme: Treatment and management of an uncommon manifestation of disease. J. Neurosurg. Spine 2012, 17, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Kalokhe, G.; Grimm, S.A.; Chandler, J.P.; Helenowski, I.; Rademaker, A.; Raizer, J.J. Metastatic glioblastoma: Case presentations and a review of the literature. J. Neurooncol. 2012, 107, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Roselli, A.; Hayry, V.; Tynninen, O.; Wartiovaara, K.; Korja, M.; Nordfors, K.; Haapasalo, H.; Nupponen, N.N. Amplification and overexpression of KIT, PDGFRA, and VEGFR2 in medulloblastomas and primitive neuroectodermal tumors. J. Neurooncol. 2010, 97, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Holand, K.; Salm, F.; Arcaro, A. The phosphoinositide 3-kinase signaling pathway as a therapeutic target in grade IV brain tumors. Curr. Cancer Drug Targets 2011, 11, 894–918. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Werb, Z. Matrix metalloproteinases: Effectors of development and normal physiology. Genes Dev. 2000, 14, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Wang, Q.; Fu, X.H.; Huang, X.H.; Chen, X.L.; Cao, L.Q.; Chen, L.Z.; Tan, H.X.; Li, W.; Bi, J.; et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol. Res. 2009, 39, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Li, H.; Liu, Y.; Guo, A.; Xu, X.; Qu, X.; Wang, S.; Zhao, J.; Li, Y.; Cao, Y. Resveratrol Inhibits the Invasion of Glioblastoma-Initiating Cells via Down-Regulation of the PI3K/Akt/NF-κB Signaling Pathway. Nutrients 2015, 7, 4383–4402. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Greenspoon, J.N.; Sharieff, W.; Hirte, H.; Overholt, A.; Devillers, R.; Gunnarsson, T.; Whitton, A. Fractionated stereotactic radiosurgery with concurrent temozolomide chemotherapy for locally recurrent glioblastoma multiforme: A prospective cohort study. Onco Targets Ther. 2014, 7, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Merrill, M.J.; Edwards, N.A. Insulin-like growth factor-I receptors in human glial tumors. J. Clin. Endocrinol. Metab. 1990, 71, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Koul, N.; Dixit, D.; Sharma, V.; Sen, E. IGF-1 induced HIF-1alpha-TLR9 cross talk regulates inflammatory responses in glioma. Cell Signal. 2011, 23, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.C.; Hung, C.M.; Chen, Y.; Liu, L.C.; Chang, T.C.; Kao, J.Y.; Ho, C.T.; Way, T.D. Osthole inhibits insulin-like growth factor-1-induced epithelial to mesenchymal transition via the inhibition of PI3K/Akt signaling pathway in human brain cancer cells. J. Agric. Food Chem. 2014, 62, 5061–5071. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Girnita, A.; Stromberg, T.; Khan, Z.; Andersson, S.; Zheng, H.; Ericsson, C.; Axelson, M.; Nister, M.; Larsson, O.; et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro Oncol. 2010, 12, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Martens, T.; Schmidt, N.O.; Eckerich, C.; Fillbrandt, R.; Merchant, M.; Schwall, R.; Westphal, M.; Lamszus, K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin. Cancer Res. 2006, 12, 6144–6152. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Kongkham, P.N.; Onvani, S.; Smith, C.A.; Rutka, J.T. Inhibition of the MET Receptor Tyrosine Kinase as a Novel Therapeutic Strategy in Medulloblastoma. Transl. Oncol. 2010, 3, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Yang, X.; Lv, S.; Wang, L.; Fan, K.; Shi, R.; Wang, F.; Song, H.; Ma, X.; Tan, X.; et al. CXCR4 Signaling Induced Epithelial-Mesenchymal Transition by PI3K/AKT and ERK Pathways in Glioblastoma. Mol. Neurobiol. 2015, 52, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Kil, W.J.; Tofilon, P.J.; Camphausen, K. Post-radiation increase in VEGF enhances glioma cell motility in vitro. Radiat. Oncol. 2012, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.A.; Karpel-Massler, G.; Bruhl, O.; Enzenmuller, S.; La Ferla-Bruhl, K.; Siegelin, M.D.; Nonnenmacher, L.; Debatin, K.M. A critical evaluation of PI3K inhibition in Glioblastoma and Neuroblastoma therapy. Mol. Cell. Ther. 2014, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Ibaragi, S.; Hu, G.F. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009, 69, 7135–7139. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Berens, M.E.; Giese, A. “...those left behind.” Biology and oncology of invasive glioma cells. Neoplasia 1999, 1, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; Moenner, M.; Javerzat, S.; North, S.; Hagedorn, M. Inhibition of angiogenesis and the angiogenesis/invasion shift. Biochem. Soc. Trans. 2011, 39, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Creasy, C.L. EZH2 as a potential target in cancer therapy. Epigenomics 2014, 6, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Prezioso, C.; Orlando, V. Polycomb proteins in mammalian cell differentiation and plasticity. FEBS Lett. 2011, 585, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Orzan, F.; Pellegatta, S.; Poliani, P.L.; Pisati, F.; Caldera, V.; Menghi, F.; Kapetis, D.; Marras, C.; Schiffer, D.; Finocchiaro, G. Enhancer of Zeste 2 (EZH2) is up-regulated in malignant gliomas and in glioma stem-like cells. Neuropathol. Appl. Neurobiol. 2011, 37, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Tsang, D.P.; Cheng, A.S. Epigenetic regulation of signaling pathways in cancer: Role of the histone methyltransferase EZH2. J. Gastroenterol. Hepatol. 2011, 26, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; De Raedt, T.; Guney, I.; Strochlic, D.E.; Macconaill, L.E.; Beroukhim, R.; et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Wagener, N.; Holland, D.; Bulkescher, J.; Crnkovic-Mertens, I.; Hoppe-Seyler, K.; Zentgraf, H.; Pritsch, M.; Buse, S.; Pfitzenmaier, J.; Haferkamp, A.; et al. The enhancer of zeste homolog 2 gene contributes to cell proliferation and apoptosis resistance in renal cell carcinoma cells. Int. J. Cancer 2008, 123, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Fornaro, L.; Bocci, G.; Sun, L.; Farrar, W.L.; Falcone, A.; Danesi, R. EZH2 inhibition: Targeting the crossroad of tumor invasion and angiogenesis. Cancer Metastasis Rev. 2012, 31, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Litzenburger, U.M.; Sahm, F.; Rauschenbach, K.J.; Tudoran, R.; Hartmann, C.; Marquez, V.E.; von Deimling, A.; Wick, W.; Platten, M. Promotion of glioblastoma cell motility by enhancer of zeste homolog 2 (EZH2) is mediated by AXL receptor kinase. PLoS ONE 2012, 7, e47663. [Google Scholar] [CrossRef] [PubMed]
- Avilla, E.; Guarino, V.; Visciano, C.; Liotti, F.; Svelto, M.; Krishnamoorthy, G.; Franco, R.; Melillo, R.M. Activation of TYRO3/AXL tyrosine kinase receptors in thyroid cancer. Cancer Res. 2011, 71, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gong, M.; Li, X.; Zhou, Y.; Gao, W.; Tulpule, A.; Chaudhary, P.M.; Jung, J.; Gill, P.S. Induction, regulation, and biologic function of Axl receptor tyrosine kinase in Kaposi sarcoma. Blood 2010, 116, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, H.; Logsdon, C.D.; Rashid, A.; Fleming, J.B.; Abbruzzese, J.L.; Gomez, H.F.; Evans, D.B.; Wang, H. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer 2011, 117, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.M.; Keating, A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014, 1542, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Staflin, K.; Zuchner, T.; Honeth, G.; Darabi, A.; Lundberg, C. Identification of proteins involved in neural progenitor cell targeting of gliomas. BMC Cancer 2009, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Vajkoczy, P.; Knyazev, P.; Kunkel, A.; Capelle, H.H.; Behrndt, S.; von Tengg-Kobligk, H.; Kiessling, F.; Eichelsbacher, U.; Essig, M.; Read, T.A.; et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. USA 2006, 103, 5799–5804. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; An, Q.; Birt, M.; Pilkington, G.J.; Hafizi, S. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget 2015, 6, 16183–16197. [Google Scholar] [CrossRef] [PubMed]
- Gely-Pernot, A.; Coronas, V.; Harnois, T.; Prestoz, L.; Mandairon, N.; Didier, A.; Berjeaud, J.M.; Monvoisin, A.; Bourmeyster, N.; De Frutos, P.G.; et al. An endogenous vitamin K-dependent mechanism regulates cell proliferation in the brain subventricular stem cell niche. Stem Cells 2012, 30, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Meng, L.; Jiang, X.; Cvm, N.K.; Ding, J.; Li, Q.; Lu, Q. TAM receptors support neural stem cell survival, proliferation and neuronal differentiation. PLoS ONE 2014, 9, e115140. [Google Scholar] [CrossRef] [PubMed]
- Weinger, J.G.; Brosnan, C.F.; Loudig, O.; Goldberg, M.F.; Macian, F.; Arnett, H.A.; Prieto, A.L.; Tsiperson, V.; Shafit-Zagardo, B. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2011, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Naganuma, H.; Satoh, E.; Nagasaka, M.; Isoe, S.; Nakano, S.; Nukui, H. Secretion of transforming growth factor-β 1 and -β 2 by malignant glioma cells. Neurol. Medico Chir. (Tokyo) 1995, 35, 423–430. [Google Scholar] [CrossRef]
- Rao, Z.Y.; Cai, M.Y.; Yang, G.F.; He, L.R.; Mai, S.J.; Hua, W.F.; Liao, Y.J.; Deng, H.X.; Chen, Y.C.; Guan, X.Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Balasubramaniyan, V.; Walenkamp, A.; Kruyt, F.A. TGF-β as a therapeutic target in high grade gliomas—Promises and challenges. Biochem. Pharmacol. 2013, 85, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J. Escaping from the TGFbeta anti-proliferative control. Carcinogenesis 2006, 27, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-β in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001, 52, 401–410. [Google Scholar] [CrossRef]
- Teodorczyk, M.; Martin-Villalba, A. Sensing invasion: Cell surface receptors driving spreading of glioblastoma. J. Cell. Physiol. 2010, 222, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.Y.; Chuang, H.M.; Huang, M.H.; Lin, S.Z.; Chiou, T.W.; Harn, H.J. n-Butylidenephthalide Regulated Tumor Stem Cell Genes EZH2/AXL and Reduced Its Migration and Invasion in Glioblastoma. Int. J. Mol. Sci. 2017, 18, 372. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kandalam, M.M.; Beta, M.; Maheswari, U.K.; Swaminathan, S.; Krishnakumar, S. Oncogenic microRNA 17-92 cluster is regulated by epithelial cell adhesion molecule and could be a potential therapeutic target in retinoblastoma. Mol. Vis. 2012, 18, 2279–2287. [Google Scholar] [PubMed]
- Chang, C.C.; Yang, Y.J.; Li, Y.J.; Chen, S.T.; Lin, B.R.; Wu, T.S.; Lin, S.K.; Kuo, M.Y.; Tan, C.T. MicroRNA-17/20a functions to inhibit cell migration and can be used a prognostic marker in oral squamous cell carcinoma. Oral Oncol. 2013, 49, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tang, H. MicroRNAs regulate the epithelial to mesenchymal transition (EMT) in cancer progression. Microrna 2014, 3, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Lopez, A.; Moreno-Bueno, G.; Cano, A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 2014, 6, 205–216. [Google Scholar] [PubMed]
- Li, Q.; Cheng, Q.; Chen, Z.; Peng, R.; Chen, R.; Ma, Z.; Wan, X.; Liu, J.; Meng, M.; Peng, Z.; et al. MicroRNA-663 inhibits the proliferation, migration and invasion of glioblastoma cells via targeting TGF-beta1. Oncol. Rep. 2016, 35, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Perdigao-Henriques, R.; Petrocca, F.; Altschuler, G.; Thomas, M.P.; Le, M.T.; Tan, S.M.; Hide, W.; Lieberman, J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene 2016, 35, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Beland, F.A.; Pogribny, I.P. E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int. J. Cancer 2010, 126, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Sole-Padulles, C.; Castro-Fornieles, J.; de la Serna, E.; Romero, S.; Calvo, A.; Sanchez-Gistau, V.; Padros-Fornieles, M.; Baeza, I.; Bargallo, N.; Frangou, S.; et al. Altered Cortico-Striatal Connectivity in Offspring of Schizophrenia Patients Relative to Offspring of Bipolar Patients and Controls. PLoS ONE 2016, 11, e0148045. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Zheng, Y.; Zhao, Y.; Xiu, X.; Wang, J. miR-590-3p suppresses cancer cell migration, invasion and epithelial-mesenchymal transition in glioblastoma multiforme by targeting ZEB1 and ZEB2. Biochem. Biophys. Res. Commun. 2015, 468, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tao, T.; Yan, W.; Feng, Y.; Wang, Y.; Cai, J.; You, Y.; Jiang, T.; Jiang, C. Upregulation of miR-181s reverses mesenchymal transition by targeting KPNA4 in glioblastoma. Sci. Rep. 2015, 5, 13072. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wang, R.; Fang, J.; Ji, X.; Li, J.; Chen, X.; Sun, G.; Wang, Z.; Liu, W.; Wang, Y.; et al. MiR-154 Functions as a Tumor Suppressor in Glioblastoma by Targeting Wnt5a. Mol. Neurobiol. 2017, 54, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.J.; Qi, Z.X.; Chen, L.C.; Yao, Y.; Gong, Y.; Mao, Y. miR-124 suppresses the migration and invasion of glioma cells in vitro via Capn4. Oncol. Rep. 2016, 35, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Bai, Y.; Qiu, S.; Zheng, L.; Huang, L.; Liu, T.; Wang, X.; Liu, Y.; Xu, N.; Yan, X.; et al. MiR-203 downregulation is responsible for chemoresistance in human glioblastoma by promoting epithelial-mesenchymal transition via SNAI2. Oncotarget 2015, 6, 8914–8928. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Greening, D.W.; Zhu, H.J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Abd Elmageed, Z.Y.; Yang, Y.; Thomas, R.; Ranjan, M.; Mondal, D.; Moroz, K.; Fang, Z.; Rezk, B.M.; Moparty, K.; Sikka, S.C.; et al. Neoplastic reprogramming of patient-derived adipose stem cells by prostate cancer cell-associated exosomes. Stem Cells 2014, 32, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Lee, H.; Weissleder, R. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Arscott, W.T.; Tandle, A.T.; Zhao, S.; Shabason, J.E.; Gordon, I.K.; Schlaff, C.D.; Zhang, G.; Tofilon, P.J.; Camphausen, K.A. Ionizing radiation and glioblastoma exosomes: Implications in tumor biology and cell migration. Transl. Oncol. 2013, 6, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Kiefel, H.; Bondong, S.; Hazin, J.; Ridinger, J.; Schirmer, U.; Riedle, S.; Altevogt, P. L1CAM: A major driver for tumor cell invasion and motility. Cell Adhes. Migr. 2012, 6, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, Y.; Chilukuri, K.; Brady, O.A.; Boulos, M.I.; Kappes, J.C.; Galileo, D.S. L1 stimulation of human glioma cell motility correlates with FAK activation. J. Neurooncol. 2011, 105, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Gast, D.; Riedle, S.; Kiefel, H.; Muerkoster, S.S.; Schafer, H.; Schafer, M.K.; Altevogt, P. The RGD integrin binding site in human L1-CAM is important for nuclear signaling. Exp. Cell Res. 2008, 314, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, V.; Temburni, M.K.; Kappes, J.C.; Galileo, D.S. L1CAM stimulates glioma cell motility and proliferation through the fibroblast growth factor receptor. Clin. Exp. Metastasis 2013, 30, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Kiefel, H.; Bondong, S.; Pfeifer, M.; Schirmer, U.; Erbe-Hoffmann, N.; Schafer, H.; Sebens, S.; Altevogt, P. EMT-associated up-regulation of L1CAM provides insights into L1CAM-mediated integrin signalling and NF-kappaB activation. Carcinogenesis 2012, 33, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Chekhonin, V.P. Extracellular vesicles shed by glioma cells: Pathogenic role and clinical value. Tumour Biol. 2014, 35, 8425–8438. [Google Scholar] [CrossRef] [PubMed]
- Van den Boorn, J.G.; Dassler, J.; Coch, C.; Schlee, M.; Hartmann, G. Exosomes as nucleic acid nanocarriers. Adv. Drug Deliv. Rev. 2013, 65, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2890. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, L.; Cocce, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Vigano, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.M.; Hossain, A.; Gumin, J.; Momin, E.N.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Parker-Kerrigan, B.; Fueyo, J.; et al. Mesenchymal Stem Cells as Natural Bio-Factories for Exosomes Carrying miR-124a in the Treatment of Gliomas. Neuro Oncol. 2017, 20, 380–390. [Google Scholar] [CrossRef] [PubMed]







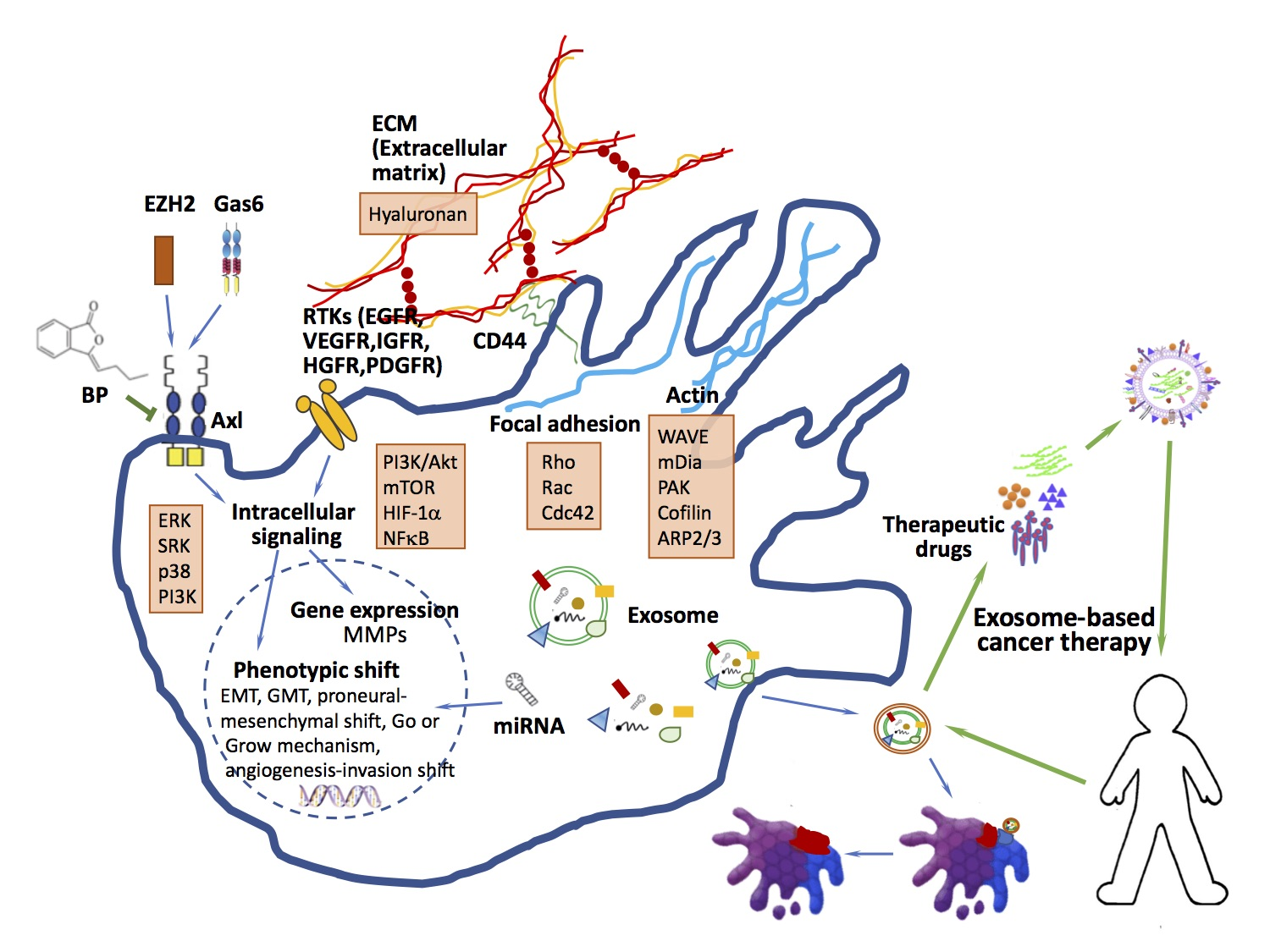{kind=link}
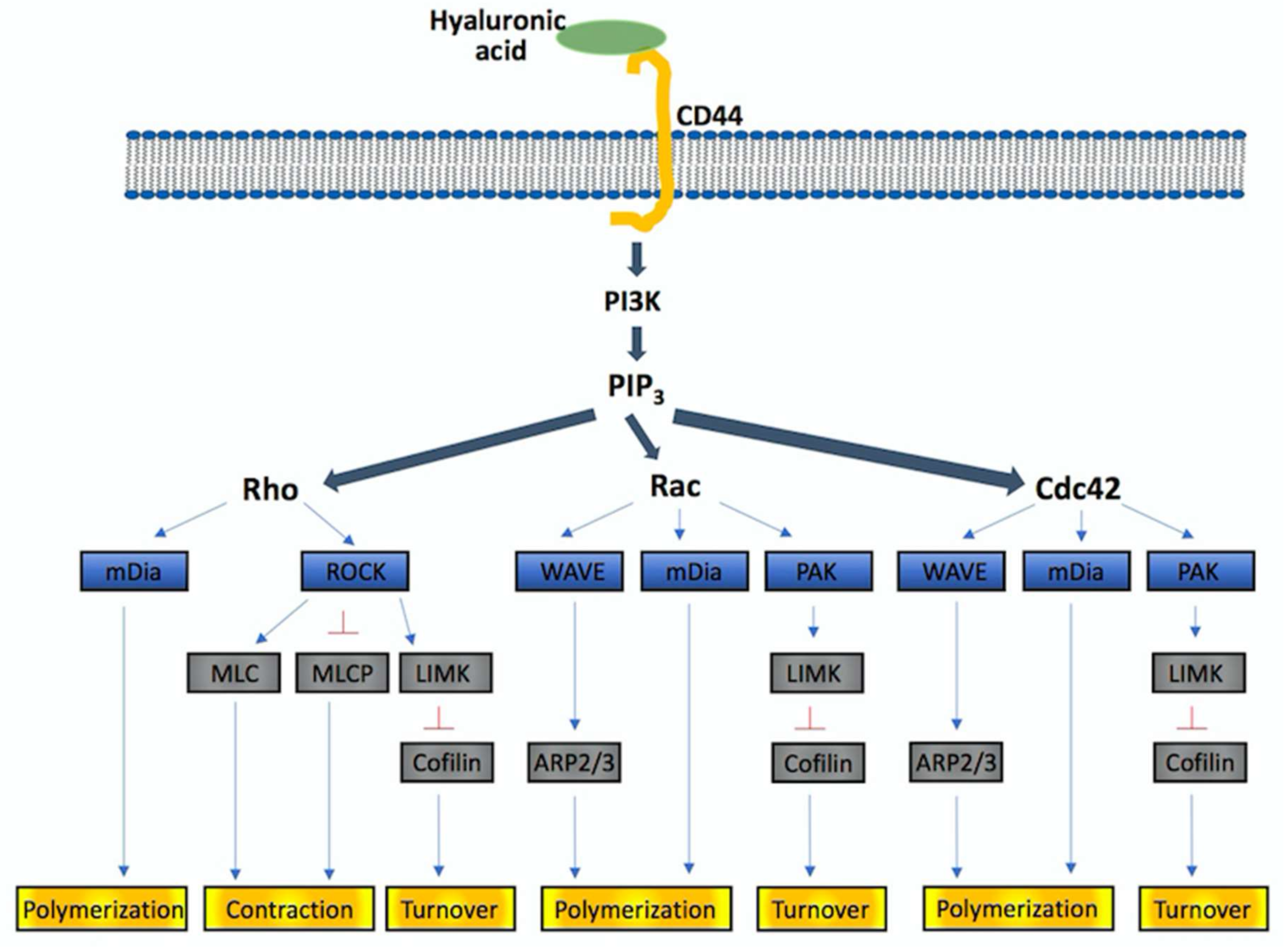{kind=link}
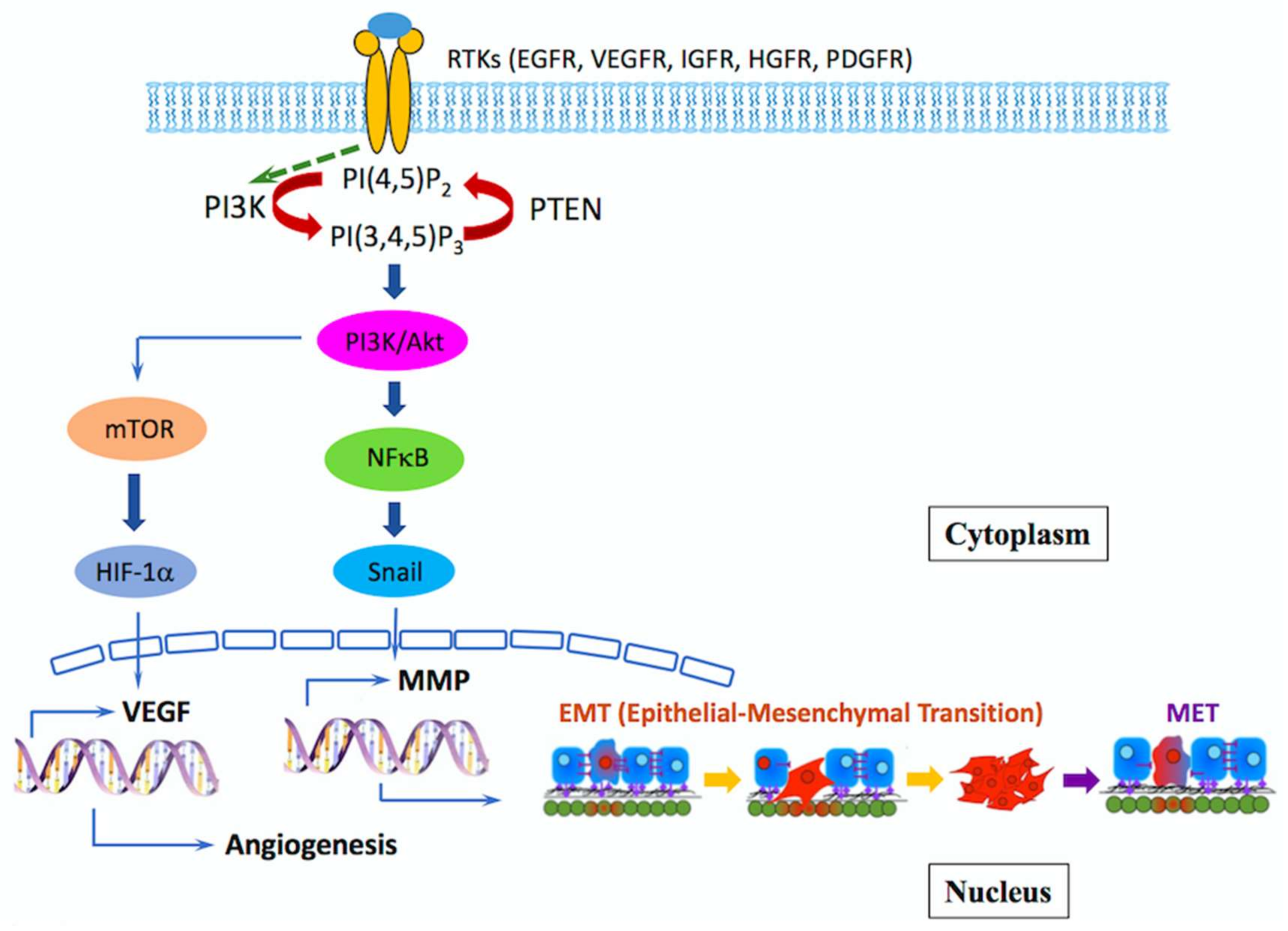{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Proneural (n = 37) | Neural (n = 19) | Classical (n = 22) | Mesenchymal (n = 38) |
|---|---|---|---|---|
| TP53 | 20 (54%) * | 4 (21%) | 0 (0%) | 12 (32%) |
| PTEN | 6 (16%) | 4 (21%) | 5 (23%) | 12 (32%) |
| NF1 | 2 (5%) | 3 (16%) | 1 (5%) | 14 (37%) * |
| EGFR | 6 (16%) | 5 (26%) | 7 (32%) | 2 (5%) |
| IDH1 | 11 (30%) ** | 1 (5%) | 0 (0%) | 0 (0%) |
| EGFRvIII | 1 (3%) | 0 (0%) | 5 (23%) | 1 (3%) |
| PDGFRA | 4 (11%) | 0 (0%) | 0 (0%) | 0 (0%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.-A.; Chang, C.-Y.; Hsueh, K.-W.; Su, H.-L.; Chiou, T.-W.; Lin, S.-Z.; Harn, H.-J. Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment. Int. J. Mol. Sci. 2018, 19, 1115. https://doi.org/10.3390/ijms19041115
Liu C-A, Chang C-Y, Hsueh K-W, Su H-L, Chiou T-W, Lin S-Z, Harn H-J. Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment. International Journal of Molecular Sciences. 2018; 19(4):1115. https://doi.org/10.3390/ijms19041115
Chicago/Turabian StyleLiu, Ching-Ann, Chia-Yu Chang, Kuo-Wei Hsueh, Hong-Lin Su, Tzyy-Wen Chiou, Shinn-Zong Lin, and Horng-Jyh Harn. 2018. "Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment" International Journal of Molecular Sciences 19, no. 4: 1115. https://doi.org/10.3390/ijms19041115
APA StyleLiu, C. -A., Chang, C. -Y., Hsueh, K. -W., Su, H. -L., Chiou, T. -W., Lin, S. -Z., & Harn, H. -J. (2018). Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment. International Journal of Molecular Sciences, 19(4), 1115. https://doi.org/10.3390/ijms19041115