The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding

 , , ,
, , ,  and
and
Abstract
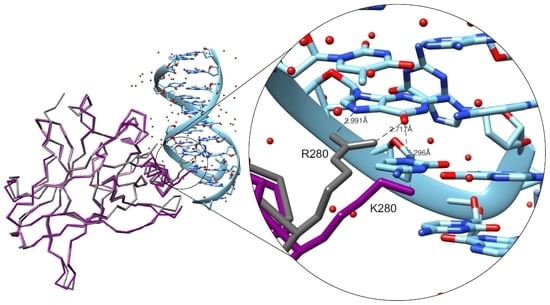
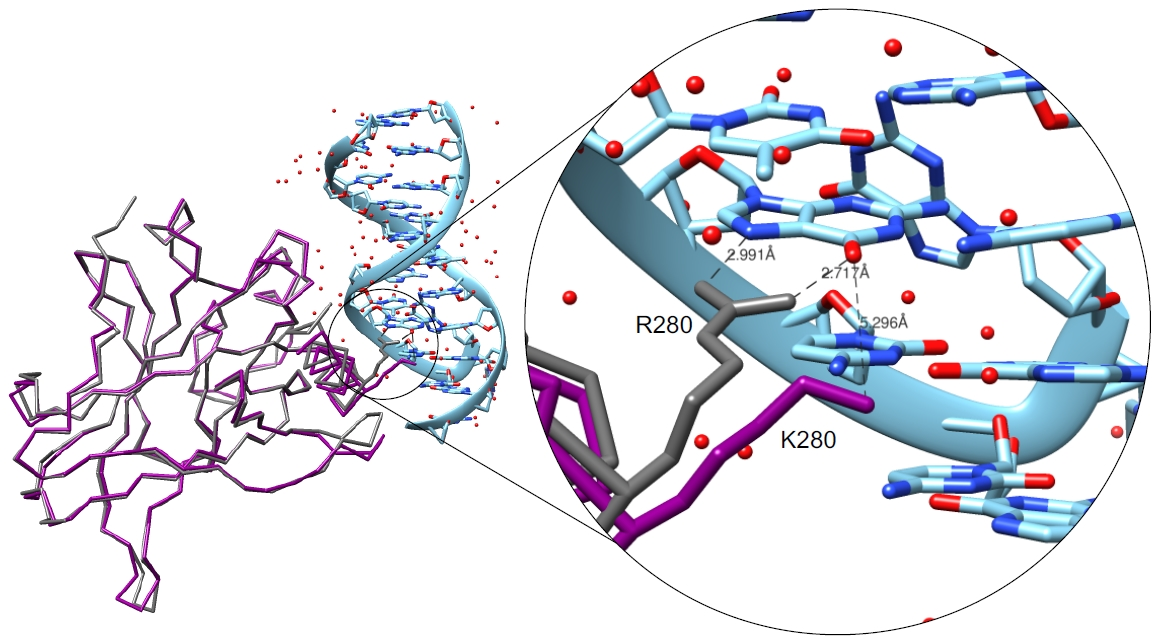
:
1. Introduction
2. Results
2.1. Expression and Purification of Mutant p53R280K DBD
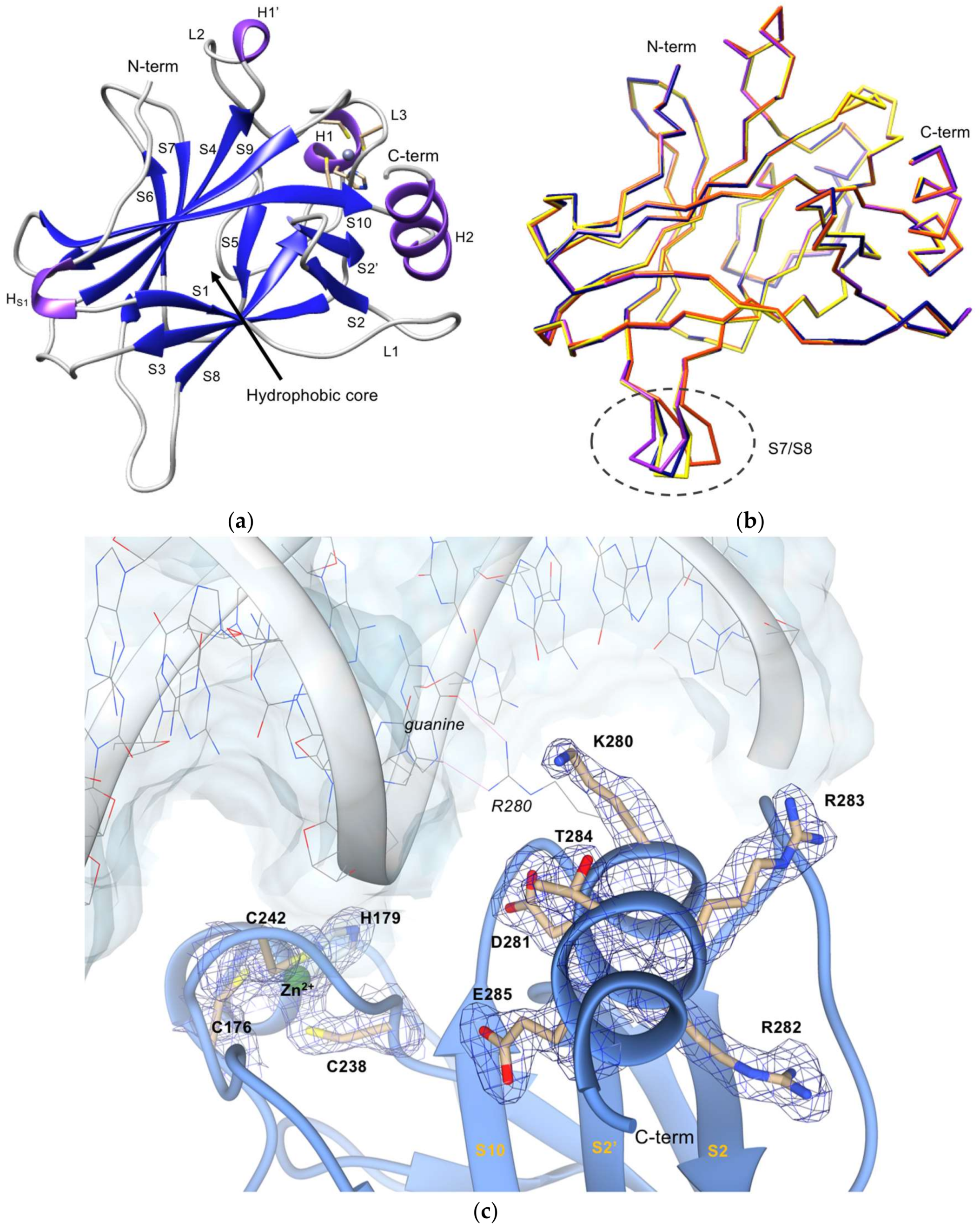
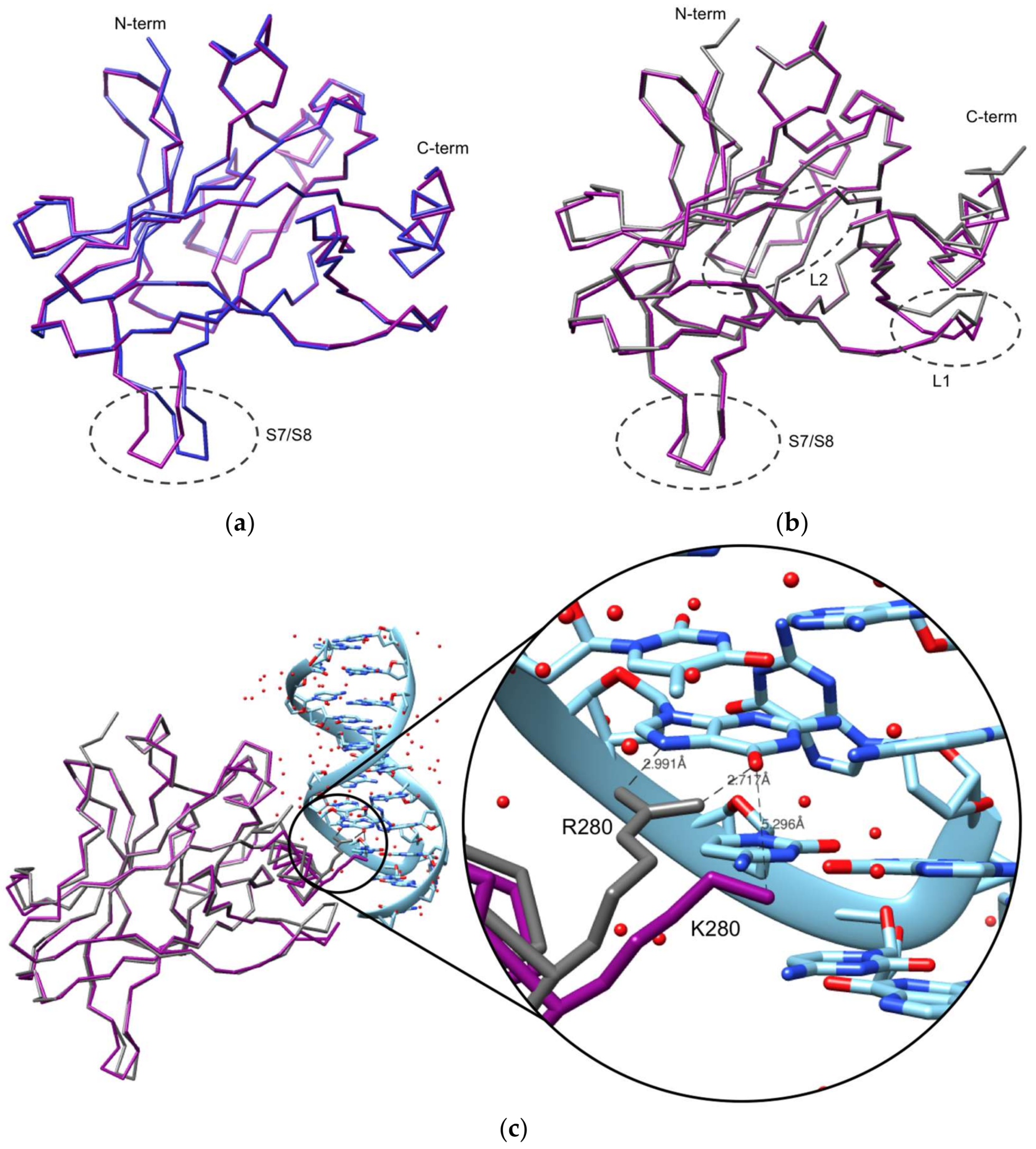
2.2. Crystallization and Structural Elucidation of Mutant p53R280K DBD
3. Discussion
4. Materials and Methods
4.1. Expression Plasmid Construction of Mutant p53R280K DBD
4.2. Recombinant Production and Purification of Mutant p53R280K DBD
4.3. Differential Scanning Fluorimetry (DSF) Screening for Buffer Optimization
4.4. Crystallization of p53 R280K DBD
4.5. Data Collection, Structure Solution, and Refinement
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 pathway: Origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.D.; Noskov, S.Y.; Lim, C. Factors governing loss and rescue of DNA binding upon single and double mutations in the p53 core domain. Nucleic Acids Res. 2002, 30, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Leao, M.; Raimundo, L.; Ramos, H.; Soares, J.; Saraiva, L. P53 family interactions and yeast: Together in anticancer therapy. Drug Discov. Today 2016, 21, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in cancer: Accumulation, gain-of-function, and therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Meplan, C.; Richard, M.J.; Hainaut, P. Metalloregulation of the tumor suppressor protein p53: Zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 2000, 19, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Verhaegh, G.W.; Parat, M.O.; Richard, M.J.; Hainaut, P. Modulation of p53 protein conformation and DNA-binding activity by intracellular chelation of zinc. Mol. Carcinog. 1998, 21, 205–214. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic metallochaperone zmc1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Trisciuoglio, D.; Porru, M.; Leonetti, C.; Stoppacciaro, A.; D’Orazi, V.; Avantaggiati, M.; Crispini, A.; Pucci, D.; D’Orazi, G. A fluorescent curcumin-based Zn(ii)-complex reactivates mutant (r175h and r273h) p53 in cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 72. [Google Scholar] [CrossRef] [PubMed]
- IARC_TP53_Database. Available online: http://www-p53.iarc.fr/ (accessed on 6 April 2018).
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of tp53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. Tp53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Arlt, C.; Ihling, C.H.; Sinz, A. Structure of full-length p53 tumor suppressor probed by chemical cross-linking and mass spectrometry. Proteomics 2015, 15, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, P.V.; Henckel, J.; Lane, D.P.; Fersht, A.R. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. USA 1998, 95, 14675–14680. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. P53 proteoforms and intrinsic disorder: An illustration of the protein structure-function continuum concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered n-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Tichy, B.; Damborsky, J.; Kabathova, J.; Trbusek, M.; Mayer, J.; Pospisilova, S. Analysis of the DNA-binding activity of p53 mutants using functional protein microarrays and its relationship to transcriptional activation. Biol. Chem. 2010, 391, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Shin, J.M.; Park, H.J.; Jang, H.O.; Bae, M.K.; Bae, S.K. Gain-of-function mutant p53-r280k mediates survival of breast cancer cells. Genes Genom. 2014, 36, 171–178. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum udpase entpd5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Zhang, L.; He, Q.Y. Fractionation of proteins by heparin chromatography. Methods Mol. Biol. 2008, 424, 213–221. [Google Scholar] [PubMed]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Allen, M.D.; Fersht, A.R. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 2004, 279, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Rozenberg, H.; Diskin-Posner, Y.; Rohs, R.; Shakked, Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013, 41, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Ang, H.C.; Veprintsev, D.B.; Blair, C.M.; Fersht, A.R. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005, 280, 16030–16037. [Google Scholar] [CrossRef] [PubMed]
- Suad, O.; Rozenberg, H.; Brosh, R.; Diskin-Posner, Y.; Kessler, N.; Shimon, L.J.; Frolow, F.; Liran, A.; Rotter, V.; Shakked, Z. Structural basis of restoring sequence-specific DNA binding and transactivation to mutant p53 by suppressor mutations. J. Mol. Biol. 2009, 385, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Chai, X.; Johnston, K.; Clements, A.; Marmorstein, R. Crystal structure of the mouse p53 core DNA-binding domain at 2.7 a resolution. J. Biol. Chem. 2001, 276, 12120–12127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rosengarth, A.; Luecke, H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Sokalingam, S.; Raghunathan, G.; Soundrarajan, N.; Lee, S.G. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS ONE 2012, 7, e40410. [Google Scholar] [CrossRef] [PubMed]
- Ishioka, C.; Frebourg, T.; Yan, Y.X.; Vidal, M.; Friend, S.H.; Schmidt, S.; Iggo, R. Screening patients for heterozygous p53 mutations using a functional assay in yeast. Nat. Genet. 1993, 5, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the ccp4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The pdb_redo server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Data Collection and Processing | |
|---|---|
| X-ray source | ESRF, ID30B |
| Wavelength (Å) | 0.9677 |
| Space group | P 1 21 1 |
| Unit-cell parameters (Å, °) | a = 68.6, b = 69.4, c = 83.3, β = 90.04 |
| Resolution range (Å) | 41.67–2.0 (2.07–2.0) |
| Solvent content (%) | 39 |
| Protein molecules per asymmetric unit | 4 |
| Matthews coefficient (Å3.Da−1) | 2.01 |
| Mosaicity (°) | 0.33 |
| I/σ (I) | 8.6 (2.0) |
| Wilson B-factor | 26.1 |
| Rmerge† (%) | 0.118 (0.833) |
| Rp.i.m.+ (%) | 0.069 (0.475) |
| Half-dataset correlation CC1/2 | 0.994 (0.815) |
| Multiplicity | 3.9 (4.0) |
| Total reflections | 197358 (14904) |
| Unique reflections | 51674 (5153) |
| Completeness (%) | 97.3 (97.2) |
| Anomalous completeness (%) | 93.2 (93.6) |
| Anomalous multiplicity | 1.9 (2.0) |
| Refinement statistics | |
| Protein atoms | 6073 |
| Zinc ions | 4 |
| Water molecules | 336 |
| Rwork‡ (%) | 0.194 |
| Rfree§ (%) | 0.237 |
| Root-mean-square deviation (r.m.s.d.) bond lengths (Å) | 0.019 |
| R.m.s.d. bond angles (°) | 1.93 |
| Average B-factor (Å2) | 31.0 |
| Protein | |
| Main-chain (A, B, C, D) | 29.1, 30.2, 28.8, 28.3 |
| Side-chain (A, B, C, D) | 33.1, 34.8, 32.3, 32.8 |
| Zinc ions (A, B, C, D) | 30.5, 24.5, 25.0, 32.1 (occ 1.0) |
| Water molecules | 30.9 |
| Ramachandran plot | |
| Residues in favoured regions (%) | 99.7 |
| Residues in allowed regions (%) | 0.26 |
| Residues outliers (%) | 0.0 |
| PDB (Protein Data Bank) accession code | 6FF9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, A.S.; Trovão, F.; Andrade Pinheiro, B.; Freire, F.; Gomes, S.; Oliveira, C.; Domingues, L.; Romão, M.J.; Saraiva, L.; Carvalho, A.L. The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. Int. J. Mol. Sci. 2018, 19, 1184. https://doi.org/10.3390/ijms19041184
Gomes AS, Trovão F, Andrade Pinheiro B, Freire F, Gomes S, Oliveira C, Domingues L, Romão MJ, Saraiva L, Carvalho AL. The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. International Journal of Molecular Sciences. 2018; 19(4):1184. https://doi.org/10.3390/ijms19041184
Chicago/Turabian StyleGomes, Ana Sara, Filipa Trovão, Benedita Andrade Pinheiro, Filipe Freire, Sara Gomes, Carla Oliveira, Lucília Domingues, Maria João Romão, Lucília Saraiva, and Ana Luísa Carvalho. 2018. "The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding" International Journal of Molecular Sciences 19, no. 4: 1184. https://doi.org/10.3390/ijms19041184
APA StyleGomes, A. S., Trovão, F., Andrade Pinheiro, B., Freire, F., Gomes, S., Oliveira, C., Domingues, L., Romão, M. J., Saraiva, L., & Carvalho, A. L. (2018). The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. International Journal of Molecular Sciences, 19(4), 1184. https://doi.org/10.3390/ijms19041184