State-of-the-Art Fluorescence Fluctuation-Based Spectroscopic Techniques for the Study of Protein Aggregation



Abstract
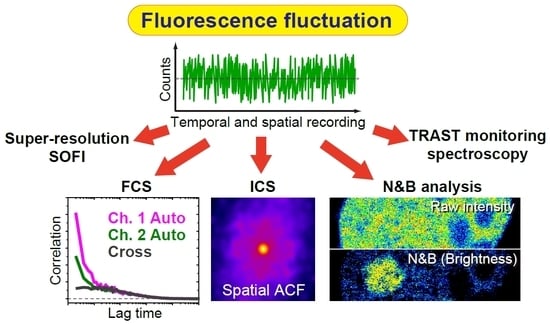
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Protein Folding and Misfolding
1.2. Transactivation Response Element (TAR) DNA/RNA-Binding Protein 43-kDa (TDP-43)
1.3. Amyloid Beta/β-Amyloid Peptide (Aβ)
1.4. Expanded Polyglutamine (polyQ) Repeat Proteins
1.5. Importance of Using Fluorescence Fluctuation
2. Fluorescence Fluctuation-Based Spectroscopic Techniques
2.1. Fluorescence Correlation Spectroscopy (FCS)
2.2. Image Correlation Spectroscopy (ICS)
2.3. Number and Brightness (N&B) Analysis and Photobleaching ICS (pbICS)
2.4. Fluctuation-Based Super-Resolution Microscopy
2.5. Transient-State Monitoring Using FCS
2.6. Transient State (TRAST) Monitoring Spectroscopy Using Time-Resolved Excitation
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FCS | Fluorescence correlation spectroscopy |
| ICS | Image correlation spectroscopy |
| pbICS | Photobleaching ICS |
| N&B | Number and brightness |
| SOFI | Super-resolution optical fluctuation imaging |
| TRAST | Transient state |
| ACF | Autocorrelation function |
| APD | Avalanche photodiode |
| PMT | Photomultiplier tube |
| LSM | Laser scanning microscopy |
| IBs | Inclusion bodies |
| ALS | Amyotrophic lateral sclerosis |
| polyQ | Polyglutamine |
References
- Hartl, F.U. Cellular homeostasis and aging. Ann. Rev. Biochem. 2016, 85, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Calamini, B.; Morimoto, R.I. Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr. Top. Med. Chem. 2012, 12, 2623–2640. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Ann. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Powers, E.T.; Balch, W.E. The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett. 2009, 583, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The ubiquitin system, autophagy, and regulated protein degradation. Ann. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Inada, N.; Kubota, H.; Matsumoto, G.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Dysregulation of the proteasome increases the toxicity of als-linked mutant sod1. Genes Cells Devoted Mol. Cell. Mech. 2014, 19, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biochem. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of sod1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. Tdp-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. Tdp-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gendron, T.F.; Xu, Y.F.; Ko, L.W.; Yen, S.H.; Petrucelli, L. Phosphorylation regulates proteasomal-mediated degradation and solubility of tar DNA binding protein-43 c-terminal fragments. Mol. Neurodegener. 2010, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Nakayama, Y.; Shibasaki, A.; Taki, A.; Yuno, S.; Takeda, K.; Yahara, M.; Tanabe, N.; Kinjo, M. Interaction of RNA with a c-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci. Rep. 2016, 6, 19230. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yokoshi, M.; Okada, H.; Kawahara, Y. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat. Commun. 2015, 6, 6183. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Vandersteen, A.; Hubin, E.; Sarroukh, R.; De Baets, G.; Schymkowitz, J.; Rousseau, F.; Subramaniam, V.; Raussens, V.; Wenschuh, H.; Wildemann, D.; et al. A comparative analysis of the aggregation behavior of amyloid-β peptide variants. FEBS Lett. 2012, 586, 4088–4093. [Google Scholar] [CrossRef] [PubMed]
- Barao, S.; Moechars, D.; Lichtenthaler, S.F.; De Strooper, B. Bace1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci. 2016, 39, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Kubota, H. Amyloid oligomers: Dynamics and toxicity in the cytosol and nucleus. FEBS J. 2010, 277, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Long, H.; Mu, Y.; Chew, L.Y. The toxicity of amyloid β oligomers. Int. J. Mol. Sci. 2012, 13, 7303–7327. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Nagata, K.; Kinjo, M. Conformational analysis of misfolded protein aggregation by fret and live-cell imaging techniques. Int. J. Mol. Sci. 2015, 16, 6076–6092. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.L.; Waddington, L.J.; Goldie, K.N. Transmission electron microscopy of amyloid fibrils. Methods Mol. Biol. 2011, 752, 197–214. [Google Scholar] [PubMed]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Bolder, S.G.; Sagis, L.M.; Venema, P.; van der Linden, E. Thioflavin t and birefringence assays to determine the conversion of proteins into fibrils. Langmuir ACS J. Surf. Coll. 2007, 23, 4144–4147. [Google Scholar] [CrossRef] [PubMed]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Skovronsky, D.M.; Zhang, B.; Kung, M.P.; Kung, H.F.; Trojanowski, J.Q.; Lee, V.M. In vivo detection of amyloid plaques in a mouse model of alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 7609–7614. [Google Scholar] [CrossRef] [PubMed]
- Jungbauer, L.M.; Yu, C.; Laxton, K.J.; LaDu, M.J. Preparation of fluorescently-labeled amyloid-β peptide assemblies: The effect of fluorophore conjugation on structure and function. J. Mol. Recognit. 2009, 22, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Ochiishi, T.; Doi, M.; Yamasaki, K.; Hirose, K.; Kitamura, A.; Urabe, T.; Hattori, N.; Kinjo, M.; Ebihara, T.; Shimura, H. Development of new fusion proteins for visualizing amyloid-β oligomers in vivo. Sci. Rep. 2016, 6, 22712. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Diamond, M.I. Polyglutamine diseases: Emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 2007, 16, R115–R123. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E. Protein aggregation and pathogenesis of Huntington’s disease: Mechanisms and correlations. Biol. Chem. 2000, 381, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Kitamura, A.; Nagata, K. Analyzing the aggregation of polyglutamine-expansion proteins and its modulation by molecular chaperones. Methods 2011, 53, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Rothlein, C.; Miettinen, M.S.; Borwankar, T.; Burger, J.; Mielke, T.; Kumke, M.U.; Ignatova, Z. Architecture of polyglutamine-containing fibrils from time-resolved fluorescence decay. J. Biol. Chem. 2014, 289, 26817–26828. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Langer, C.A.; Boteva, R.; Bottcher, U.M.; Stemp, M.J.; Schaffar, G.; Rao, B.V.; Giese, A.; Kretzschmar, H.; Siegers, K.; et al. Chaperonin tric promotes the assembly of polyq expansion proteins into nontoxic oligomers. Mol. Cell 2006, 23, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Nollen, E.A.; Kitagawa, K.; Bindokas, V.P.; Morimoto, R.I. Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 2002, 4, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Kubota, H.; Pack, C.G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. HSP70 and HSP40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.; Geller, R.; Spiess, C.; Frydman, J. The chaperonin tric controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006, 8, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, E.; Zako, T.; Muto, H.; Itoo, Y.; Sorgjerd, K.; Terada, N.; Abe, A.; Miyazawa, M.; Kitamura, A.; Kitaura, H.; et al. Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J. Biol. Chem. 2013, 288, 19958–19972. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Rigler, R.; Mets, U.; Widengren, J.; Kask, P. Fluorescence correlation spectroscopy with high count rate and low-background—Analysis of translational diffusion. Eur. Biophys. J. Biophy. 1993, 22, 169–175. [Google Scholar] [CrossRef]
- Rigler, R.; Widengren, J. Fluorescence-based monitoring of electronic state and ion exchange kinetics with FCS and related techniques: From t-jump measurements to fluorescence fluctuations. Eur. Biophys. J. EBJ 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Okamoto, Y.; Popiel, H.A.; Fujikake, N.; Toda, T.; Kinjo, M.; Nagai, Y. Detection of polyglutamine protein oligomers in cells by fluorescence correlation spectroscopy. J. Biol. Chem. 2007, 282, 24039–24048. [Google Scholar] [CrossRef] [PubMed]
- Puchalla, J.; Krantz, K.; Austin, R.; Rye, H. Burst analysis spectroscopy: A versatile single-particle approach for studying distributions of protein aggregates and fluorescent assemblies. Proc. Natl. Acad. Sci. USA 2008, 105, 14400–14405. [Google Scholar] [CrossRef] [PubMed]
- Oura, M.; Yamamoto, J.; Ishikawa, H.; Mikuni, S.; Fukushima, R.; Kinjo, M. Polarization-dependent fluorescence correlation spectroscopy for studying structural properties of proteins in living cell. Sci. Rep. 2016, 6, 31091. [Google Scholar] [CrossRef] [PubMed]
- Mueller, V.; Honigmann, A.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Eggeling, C. FCS in STED microscopy: Studying the nanoscale of lipid membrane dynamics. Methods Enzymol. 2013, 519, 1–38. [Google Scholar] [PubMed]
- Laurence, T.A.; Ly, S.; Bourguet, F.; Fischer, N.O.; Coleman, M.A. Fluorescence correlation spectroscopy at micromolar concentrations without optical nanoconfinement. J. Phys. Chem. B 2014, 118, 9662–9667. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.; Widengren, J. Stimulated emission depletion microscopy. Chem. Rev. 2017, 117, 7377–7427. [Google Scholar] [CrossRef] [PubMed]
- Kinkhabwala, A.A.; Yu, Z.F.; Fan, S.H.; Moerner, W.E. Fluorescence correlation spectroscopy at high concentrations using gold bowtie nanoantennas. Chem. Phys. 2012, 406, 3–8. [Google Scholar] [CrossRef]
- Tjernberg, L.O.; Pramanik, A.; Bjorling, S.; Thyberg, P.; Thyberg, J.; Nordstedt, C.; Berndt, K.D.; Terenius, L.; Rigler, R. Amyloid β-peptide polymerization studied using fluorescence correlation spectroscopy. Chem. Biol. 1999, 6, 53–62. [Google Scholar] [CrossRef]
- Tiiman, A.; Jarvet, J.; Graslund, A.; Vukojevic, V. Heterogeneity and turnover of intermediates during amyloid-β (αβ) peptide aggregation studied by fluorescence correlation spectroscopy. Biochemistry 2015, 54, 7203–7211. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Walta, S.; Cadek, C.; Richtering, W.; Willbold, D. Fluorescence correlation spectroscopy reveals a cooperative unfolding of monomeric amyloid-β 42 with a low gibbs free energy. Sci. Rep. 2017, 7, 2154. [Google Scholar] [CrossRef] [PubMed]
- Wennmalm, S.; Chmyrov, V.; Widengren, J.; Tjernberg, L. Highly sensitive FRET-FCS detects amyloid β-peptide oligomers in solution at physiological concentrations. Anal. Chem. 2015, 87, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Beam, M.; Silva, M.C.; Morimoto, R.I. Dynamic imaging by fluorescence correlation spectroscopy identifies diverse populations of polyglutamine oligomers formed in vivo. J. Biol. Chem. 2012, 287, 26136–26145. [Google Scholar] [CrossRef] [PubMed]
- Mikuni, S.; Kodama, K.; Sasaki, A.; Kohira, N.; Maki, H.; Munetomo, M.; Maenaka, K.; Kinjo, M. Screening for FTSZ dimerization inhibitors using fluorescence cross-correlation spectroscopy and surface resonance plasmon analysis. PLoS ONE 2015, 10, e0130933. [Google Scholar] [CrossRef] [PubMed]
- Serpionov, G.V.; Alexandrov, A.I.; Antonenko, Y.N.; Ter-Avanesyan, M.D. A protein polymerization cascade mediates toxicity of non-pathological human huntingtin in yeast. Sci. Rep. 2015, 5, 18407. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Yuno, S.; Muto, H.; Kinjo, M. Different aggregation states of a nuclear localization signal-tagged 25-kDa c-terminal fragment of tar RNA/DNA-binding protein 43 kDa. Genes Cells Devoted Mol. Cell. Mech. 2017, 22, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Ha, T.; Kim, H.D.; Centner, T.; Labeit, S.; Chu, S. Fluorescence quenching: A tool for single-molecule protein-folding study. Proc. Natl. Acad. Sci. USA 2000, 97, 14241–14244. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.L. A theory of sensitized luminescence in solids. J. Chem. Phys. 1953, 21, 836–850. [Google Scholar] [CrossRef]
- Murphy, C.B.; Zhang, Y.; Troxler, T.; Ferry, V.; Martin, J.J.; Jones, W.E. Probing forster and dexter energy-transfer mechanisms in fluorescent conjugated polymer chemosensors. J. Phys. Chem. B 2004, 108, 1537–1543. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar] [CrossRef]
- Bogdanov, A.M.; Acharya, A.; Titelmayer, A.V.; Mamontova, A.V.; Bravaya, K.B.; Kolomeisky, A.B.; Lukyanov, K.A.; Krylov, A.I. Turning on and off photoinduced electron transfer in fluorescent proteins by pi-stacking, halide binding, and tyr145 mutations. J. Am. Chem. Soc. 2016, 138, 4807–4817. [Google Scholar] [CrossRef] [PubMed]
- Vamosi, G.; Mucke, N.; Muller, G.; Krieger, J.W.; Curth, U.; Langowski, J.; Toth, K. EGFP oligomers as natural fluorescence and hydrodynamic standards. Sci. Rep. 2016, 6, 33022. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.; Balaji, J.; Nag, S.; Kaushalya, S.K.; Maiti, S. Protein aggregation probed by two-photon fluorescence correlation spectroscopy of native tryptophan. J. Chem. Phys. 2008, 129, 075103. [Google Scholar] [CrossRef] [PubMed]
- Kannan, B.; Har, J.Y.; Liu, P.; Maruyama, I.; Ding, J.L.; Wohland, T. Electron multiplying charge-coupled device camera based fluorescence correlation spectroscopy. Anal. Chem. 2006, 78, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Gosch, M.; Serov, A.; Anhut, T.; Lasser, T.; Rochas, A.; Besse, P.A.; Popovic, R.S.; Blom, H.; Rigler, R. Parallel single molecule detection with a fully integrated single-photon 2 × 2 cmos detector array. J. Biomed. Opt. 2004, 9, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Ng, X.W.; Bag, N.; Wohland, T. Characterization of lipid and cell membrane organization by the fluorescence correlation spectroscopy diffusion law. Chimia 2015, 69, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.W.; Singh, A.P.; Bag, N.; Garbe, C.S.; Saunders, T.E.; Langowski, J.; Wohland, T. Imaging fluorescence (cross-) correlation spectroscopy in live cells and organisms. Nat. Protoc. 2015, 10, 1948–1974. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, W.G.; Varadi, G.; Entine, G.; Podniesinski, E.; Wallace, P.K. Enhanced red and near infrared detection in flow cytometry using avalanche photodiodes. Cytom. Part A J. Int. Soc. Anal. Cytol. 2008, 73, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Su, B.; Holub, O.; Weisshart, K. Flim and fcs detection in laser-scanning microscopes: Increased efficiency by GAASP hybrid detectors. Microsc. Res. Tech. 2011, 74, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Qian, H. On the statistics of fluorescence correlation spectroscopy. BioPhys. Chem. 1990, 38, 49–57. [Google Scholar] [CrossRef]
- Widengren, J.; Mets, U.; Rigler, R. Fluorescence correlation spectroscopy of triplet-states in solution—A theoretical and experimental-study. J. Phys. Chem. 1995, 99, 13368–13379. [Google Scholar] [CrossRef]
- Yamamoto, J.; Oura, M.; Yamashita, T.; Miki, S.; Jin, T.; Haraguchi, T.; Hiraoka, Y.; Terai, H.; Kinjo, M. Rotational diffusion measurements using polarization-dependent fluorescence correlation spectroscopy based on superconducting nanowire single-photon detector. Opt. Express 2015, 23, 32633–32642. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Liu, D.; Miki, S.; Yamamoto, J.; Haraguchi, T.; Kinjo, M.; Hiraoka, Y.; Wang, Z.; Terai, H. Fluorescence correlation spectroscopy with visible-wavelength superconducting nanowire single-photon detector. Opt. Express 2014, 22, 28783–28789. [Google Scholar] [CrossRef] [PubMed]
- Kolin, D.L.; Wiseman, P.W. Advances in image correlation spectroscopy: Measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cells. Cell Biochem. Biophys. 2007, 49, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Hebert, B.; Costantino, S.; Wiseman, P.W. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 2005, 88, 3601–3614. [Google Scholar] [CrossRef] [PubMed]
- Lankova, M.; Humpolickova, J.; Vosolsobe, S.; Cit, Z.; Lacek, J.; Covan, M.; Covanova, M.; Hof, M.; Petrasek, J. Determination of dynamics of plant plasma membrane proteins with fluorescence recovery and raster image correlation spectroscopy. Microsc. Microanal. 2016, 22, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Bonor, J.; Nohe, A. Image correlation spectroscopy to define membrane dynamics. Methods Mol. Biol. 2010, 591, 353–364. [Google Scholar] [PubMed]
- Clayton, A.H.; Chattopadhyay, A. Get your kICS by measuring membrane protein dynamics. Biophys. J. 2015, 109, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.; George, S.C. Theory and practical recommendations for autocorrelation-based image correlation spectroscopy. J. Biomed. Opt. 2012, 17, 080801. [Google Scholar] [CrossRef] [PubMed]
- Vetri, V.; Ossato, G.; Militello, V.; Digman, M.A.; Leone, M.; Gratton, E. Fluctuation methods to study protein aggregation in live cells: Concanavalin a oligomers formation. Biophys. J. 2011, 100, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hosp, F.; Frottin, F.; Ge, H.; Mann, M.; Hayer-Hartl, M.; Hartl, F.U. Soluble oligomers of polyq-expanded huntingtin target a multiplicity of key cellular factors. Mol. Cell 2016, 63, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Ciccotosto, G.D.; Kozer, N.; Chow, T.T.; Chon, J.W.; Clayton, A.H. Aggregation distributions on cells determined by photobleaching image correlation spectroscopy. Biophys. J. 2013, 104, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Digman, M.A.; Wang, L.; Gratton, E.; Albanesi, J.P.; Jameson, D.M. Oligomerization state of dynamin 2 in cell membranes using tirf and number and brightness analysis. Biophys. J. 2011, 100, L15–L17. [Google Scholar] [CrossRef] [PubMed]
- James, N.G.; Digman, M.A.; Gratton, E.; Barylko, B.; Ding, X.; Albanesi, J.P.; Goldberg, M.S.; Jameson, D.M. Number and brightness analysis of LRRK2 oligomerization in live cells. Biophys. J. 2012, 102, L41–L43. [Google Scholar] [CrossRef] [PubMed]
- Unruh, J.R.; Gratton, E. Analysis of molecular concentration and brightness from fluorescence fluctuation data with an electron multiplied CCD camera. Biophys. J. 2008, 95, 5385–5398. [Google Scholar] [CrossRef] [PubMed]
- Ossato, G.; Digman, M.A.; Aiken, C.; Lukacsovich, T.; Marsh, J.L.; Gratton, E. A two-step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis. Biophys. J. 2010, 98, 3078–3085. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Kinjo, M. Determination of diffusion coefficients in live cells using fluorescence recovery after photobleaching with wide-field fluorescence microscopy. Biophys. Physicobiol. 2018, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Dertinger, T.; Colyer, R.; Iyer, G.; Weiss, S.; Enderlein, J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl. Acad. Sci. USA 2009, 106, 22287–22292. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, C.; Rodriguez, C.; Park, Y.; Cho, Y.H. Superresolution imaging with optical fluctuation using speckle patterns illumination. Sci. Rep. 2015, 5, 16525. [Google Scholar] [CrossRef] [PubMed]
- Girsault, A.; Lukes, T.; Sharipov, A.; Geissbuehler, S.; Leutenegger, M.; Vandenberg, W.; Dedecker, P.; Hofkens, J.; Lasser, T. SOFI simulation tool: A software package for simulating and testing super-resolution optical fluctuation imaging. PLoS ONE 2016, 11, e0161602. [Google Scholar] [CrossRef] [PubMed]
- Widengren, J. Fluorescence-based transient state monitoring for biomolecular spectroscopy and imaging. J. R. Soc. Interface R. Soc. 2010, 7, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Widengren, J.; Schwille, P. Characterization of photoinduced isomerization and back-isomerization of the cyanine dye CY5 by fluorescence correlation spectroscopy. J. Phys. Chem. A 2000, 104, 6416–6428. [Google Scholar] [CrossRef]
- Widengren, J.; Seidel, C.A.M. Manipulation and characterization of photo-induced transient states of merocyanine 540 by fluorescence correlation spectroscopy. Phys. Chem. Chem. Phys. 2000, 2, 3435–3441. [Google Scholar] [CrossRef]
- Liu, Y.; Lilley, D.M.J. Crystal structures of cyanine fluorophores stacked onto the end of double-stranded RNA. Biophys. J. 2017, 113, 2336–2343. [Google Scholar] [CrossRef] [PubMed]
- Noukakis, D.; Vanderauweraer, M.; Toppet, S.; Deschryver, F.C. Photophysics of a thiacarbocyanine dye in organic-solvents. J. Phys. Chem. 1995, 99, 11860–11866. [Google Scholar] [CrossRef]
- Aramendia, P.F.; Negri, R.M.; Sanroman, E. Temperature-dependence of fluorescence and photoisomerization in symmetrical carbocyanines–Influence of medium viscosity and molecular-structure. J. Phys. Chem. 1994, 98, 3165–3173. [Google Scholar] [CrossRef]
- Sanden, T.; Persson, G.; Thyberg, P.; Blom, H.; Widengren, J. Monitoring kinetics of highly environment sensitive states of fluorescent molecules by modulated excitation and time-averaged fluorescence intensity recording. Anal. Chem. 2007, 79, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Hevekerl, H.; Tornmalm, J.; Widengren, J. Fluorescence-based characterization of non-fluorescent transient states of tryptophan—Prospects for protein conformation and interaction studies. Sci. Rep. 2016, 6, 35052. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Govitrapong, P.; Sharma, S.; Muralikrishnan, D.; Shavali, S.; Pellett, L.; Schafer, R.; Albano, C.; Eken, J. Ubiquinone (coenzyme q10) and mitochondria in oxidative stress of Parkinson’s disease. Biol. Signals Recept. 2001, 10, 224–253. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.E.; Mouradian, M.M. Cytoprotective mechanisms of DJ-1 against oxidative stress through modulating ERK1/2 and ASK1 signal transduction. Redox Biol. 2018, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Tornmalm, J.; Widengren, J. Label-free monitoring of ambient oxygenation and redox conditions using the photodynamics of flavin compounds and transient state (trast) spectroscopy. Methods 2017. [Google Scholar] [CrossRef] [PubMed]
- Sanden, T.; Persson, G.; Widengren, J. Transient state imaging for microenvironmental monitoring by laser scanning microscopy. Anal. Chem. 2008, 80, 9589–9596. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.; Eaton, W.A. Protein folding studied by single-molecule fret. Curr. Opin. Struct. Biol. 2008, 18, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widengren, J.; Schweinberger, E.; Berger, S.; Seidel, C.A.M. Two new concepts to measure fluorescence resonance energy transfer via fluorescence correlation spectroscopy: Theory and experimental realizations. J. Phys. Chem. A 2001, 105, 6851–6866. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitamura, A.; Kinjo, M. State-of-the-Art Fluorescence Fluctuation-Based Spectroscopic Techniques for the Study of Protein Aggregation. Int. J. Mol. Sci. 2018, 19, 964. https://doi.org/10.3390/ijms19040964
Kitamura A, Kinjo M. State-of-the-Art Fluorescence Fluctuation-Based Spectroscopic Techniques for the Study of Protein Aggregation. International Journal of Molecular Sciences. 2018; 19(4):964. https://doi.org/10.3390/ijms19040964
Chicago/Turabian StyleKitamura, Akira, and Masataka Kinjo. 2018. "State-of-the-Art Fluorescence Fluctuation-Based Spectroscopic Techniques for the Study of Protein Aggregation" International Journal of Molecular Sciences 19, no. 4: 964. https://doi.org/10.3390/ijms19040964
APA StyleKitamura, A., & Kinjo, M. (2018). State-of-the-Art Fluorescence Fluctuation-Based Spectroscopic Techniques for the Study of Protein Aggregation. International Journal of Molecular Sciences, 19(4), 964. https://doi.org/10.3390/ijms19040964