Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression

 ,
, 
Abstract
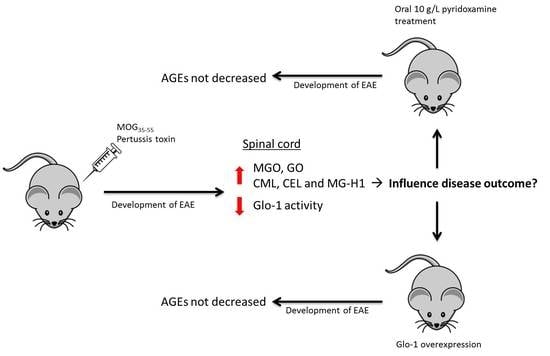
:
1. Introduction
2. Results
2.1. Experimental Autoimmune Encephalomyelitis Induces the Formation of Advanced Glycation Endproducts in the Central Nervous System
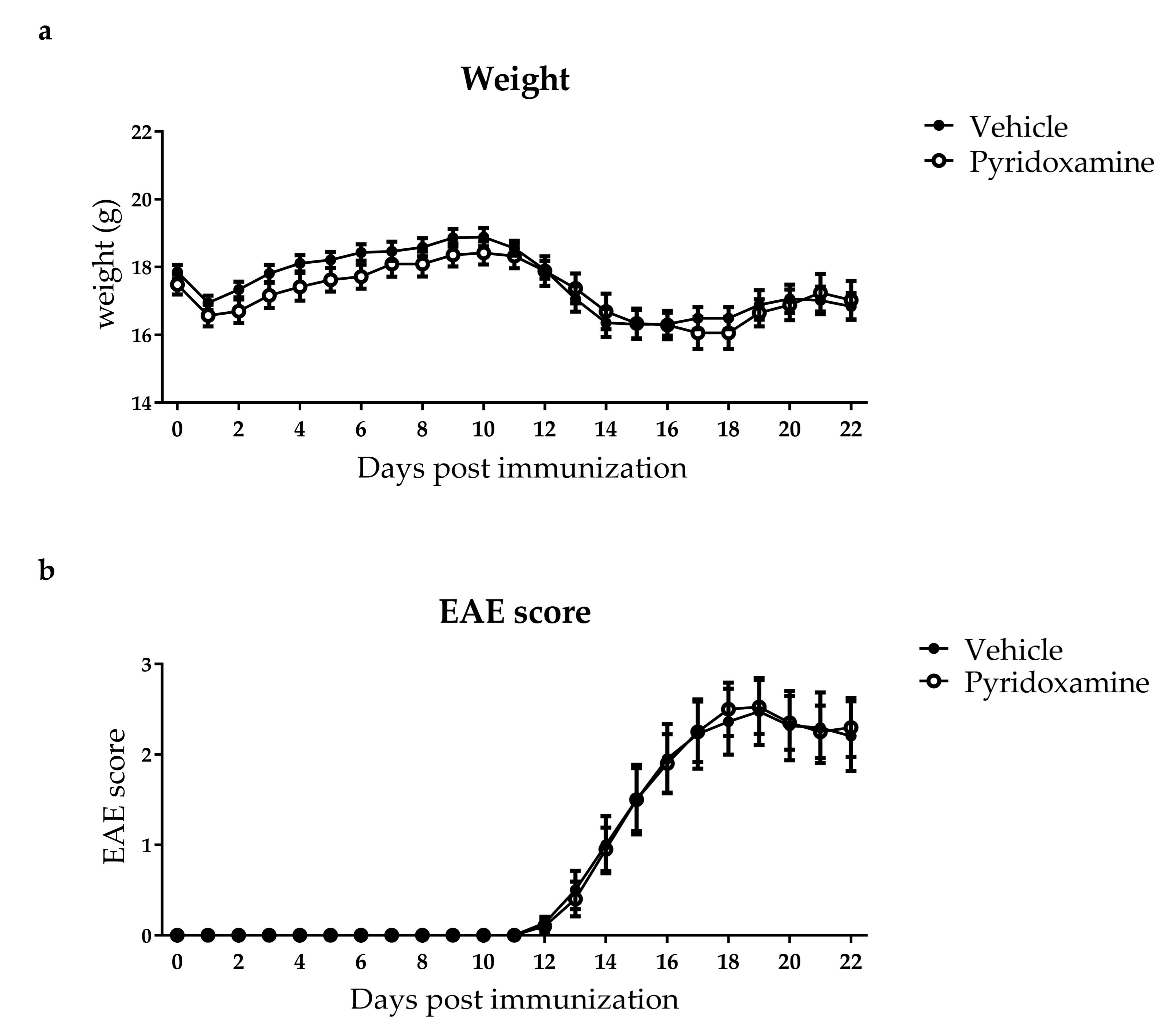
2.2. Pyridoxamine Intervention did not Reduce Advanced Glycation Endproduct Levels in the Plasma and Spinal Cord During EAE
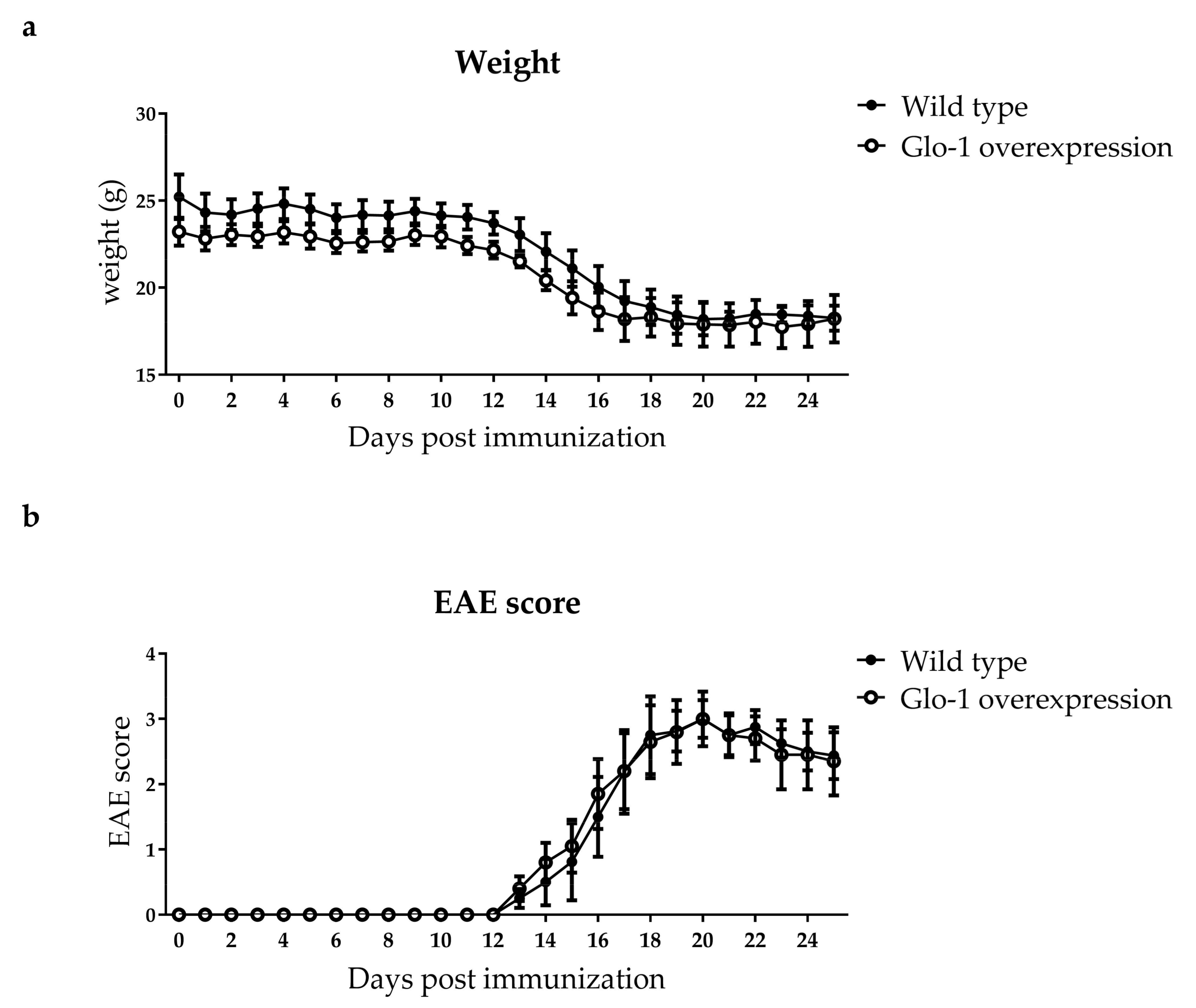
2.3. Human Glo-1 Overexpression Mouse Model did not Reduce Advanced Glycation Endproduct Levels in the Plasma and Central Nervous System during EAE
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. α-Dicarbonyl and AGE Measurements
4.3. Glyoxalase-1 Activity Assay
4.4. Glyoxalase-2 Activity Assay
4.5. Statistical Analysis
Author Contributions
Conflicts of Interest
Abbreviations
| 3DG | 3-deoxyglucosone |
| AGEs | Advanced glycation endproducts |
| CEL | Nε-(1-carboxyethyl)lysine |
| CFA | Complete freund’s adjuvant |
| CML | Nε-(carboxymethyl)lysine |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EAE | Experimental autoimmune encephalomyelitis |
| Glo-1 | Glyoxalase-1 |
| Glo-2 | Glyoxalase-2 |
| GO | Glyoxal |
| GSH | Glutathione |
| MG-H1 | Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine |
| MGO | Methylglyoxal |
| MOG | Myelin oligodendrocyte glycoprotein |
| MS | Multiple sclerosis |
| NF-κB | Nuclear factor-κB |
| RAGE | Receptor for advanced glycation endproducts |
| RR MS | Relapsing-remitting MS |
| SP MS | Secondary progressive MS |
| sRAGE | Soluble receptor for advanced glycation endproducts |
| UPLC MSMS | Ultra-performance liquid chromatography tandem mass spectrometry |
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Ellwardt, E.; Zipp, F. Molecular mechanisms linking neuroinflammation and neurodegeneration in ms. Exp. Neurol. 2014, 262, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Meyer-Rienecker, H.; Hartung, H.P.; Zettl, U.K. Animal models of multiple sclerosis--potentials and limitations. Prog. Neurobiol. 2010, 92, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of multiple sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Amor, S. Experimental autoimmune encephalomyelitis is a good model of multiple sclerosis if used wisely. Mult. Scler. Relat. Disord. 2014, 3, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial m1/m2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. Switz. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (ages and ales): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. S1), 3–27. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Van Eupen, M.G.; Schram, M.T.; Colhoun, H.M.; Hanssen, N.M.; Niessen, H.W.; Tarnow, L.; Parving, H.H.; Rossing, P.; Stehouwer, C.D.; Schalkwijk, C.G. The methylglyoxal-derived age tetrahydropyrimidine is increased in plasma of individuals with type 1 diabetes mellitus and in atherosclerotic lesions and is associated with svcam-1. Diabetologia 2013, 56, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Blaak, E.E.; et al. Nepsilon-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Niessen, P.M.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Driessen, A.; Wolfs, M.G.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of nepsilon-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassara, H. Advanced glycation end products (ages) co-localize with age receptors in the retinal vasculature of diabetic and of age-infused rats. Am. J. Pathol. 1997, 150, 523–531. [Google Scholar] [PubMed]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Munch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Dalfo, E.; Portero-Otin, M.; Ayala, V.; Martinez, A.; Pamplona, R.; Ferrer, I. Evidence of oxidative stress in the neocortex in incidental lewy body disease. J. Neuropathol. Exp. Neurol. 2005, 64, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Hennies, C.; Sternberg, D.; Wang, P.; Kinkel, P.; Hojnacki, D.; Weinstock-Guttmann, B.; Munschauer, F. Diagnostic potential of plasma carboxymethyllysine and carboxyethyllysine in multiple sclerosis. J. Neuroinflammation 2010, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Ostrow, P.; Vaughan, M.; Chichelli, T.; Munschauer, F. Age-rage in multiple sclerosis brain. Immunol. Investig. 2011, 40, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Voziyan, P.A.; Hudson, B.G. Pyridoxamine: The many virtues of a maillard reaction inhibitor. Ann. N. Y. Acad. Sci. 2005, 1043, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Wetzels, S. Department of Immunology and Biochemistry, Hasselt University, Hasselt, Belgium. Department of Internal Medicine, Maastricht University, Maastricht, the Netherlands. Glyoxalase 2 activity in the spinal cord and brain of healthy and mice subjected to EAE. 2018; Material not intended for publication. [Google Scholar]
- Bogie, J.F.; Timmermans, S.; Huynh-Thu, V.A.; Irrthum, A.; Smeets, H.J.; Gustafsson, J.A.; Steffensen, K.R.; Mulder, M.; Stinissen, P.; Hellings, N.; et al. Myelin-derived lipids modulate macrophage activity by liver x receptor activation. PLoS ONE 2012, 7, e44998. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.B.; Liggitt, D.; Goverman, J.M. Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 2014, 193, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.S.; Wu, Z.Y.; Zhang, H.P.; Furtado, G.; Chen, X.; Yan, S.F.; Schmidt, A.M.; Brown, C.; Stern, A.; LaFaille, J.; et al. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic t-cell infiltration of the central nervous system. Nat. Med. 2003, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Voziyan, P.A.; Hudson, B.G. Pyridoxamine as a multifunctional pharmaceutical: Targeting pathogenic glycation and oxidative damage. CMLS 2005, 62, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Hamm, M.W.; Mehansho, H.; Henderson, L.M. Transport and metabolism of pyridoxamine and pyridoxamine phosphate in the small intestine of the rat. J. Nutr. 1979, 109, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Asakura, T.; Mizuno, A.; Matsuda, M. Absorption and metabolism of pyridoxamine in mice. I. Pyridoxal as the only form of transport in blood. J. Nutr. Sci. Vitaminol. 1991, 37, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Asakura, T.; Mizuno, A.; Matsuda, M. Absorption and metabolism of pyridoxamine in mice. II. Transformation of pyridoxamine to pyridoxal in intestinal tissues. J. Nutr. Sci. Vitaminol. 1992, 38, 227–233. [Google Scholar] [CrossRef] [PubMed]
- van der Ham, M.; Albersen, M.; de Koning, T.J.; Visser, G.; Middendorp, A.; Bosma, M.; Verhoeven-Duif, N.M.; de Sain-van der Velden, M.G. Quantification of vitamin b6 vitamers in human cerebrospinal fluid by ultra performance liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2012, 712, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Surtees, R.; Mills, P.; Clayton, P. Inborn errors affecting vitamin b6 metabolism. Future Neurol. 2006, 1, 615–620. [Google Scholar] [CrossRef]
- Garg, S.; Syngle, A.; Vohra, K. Efficacy and tolerability of advanced glycation end-products inhibitor in osteoarthritis: A randomized, double-blind, placebo-controlled study. Clin. J. Pain 2013, 29, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Brouwers, O.; Gaens, K.H.; Wouters, K.; Cleutjens, J.P.; Janssen, B.J.; Miyata, T.; Stehouwer, C.D.; Schalkwijk, C.G. Delayed intervention with pyridoxamine improves metabolic function and prevents adipose tissue inflammation and insulin resistance in high-fat diet-induced obese mice. Diabetes 2016, 65, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Miyata, T.; Ueda, Y.; Yoshino, A.; Nangaku, M.; van Ypersele de Strihou, C.; Kurokawa, K. Efficient in vitro lowering of carbonyl stress by the glyoxalase system in conventional glucose peritoneal dialysis fluid. Kidney Int. 2002, 62, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An rna-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol. 2011, 22, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Zheng, J.; Perrone-Bizzozero, N.I.; Bizzozero, O.A. Increased carbonylation, protein aggregation and apoptosis in the spinal cord of mice with experimental autoimmune encephalomyelitis. ASN Neuro 2013, 5, e00111. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Bizzozero, O.A. Accumulation of protein carbonyls within cerebellar astrocytes in murine experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2010, 88, 3376–3385. [Google Scholar] [CrossRef] [PubMed]
- Wetzels, S.; Wouters, K.; Schalkwijk, C.G.; Vanmierlo, T.; Hendriks, J.J. Methylglyoxal-derived advanced glycation endproducts in multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 421. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Engelen, L.; Ferreira, I.; Scheijen, J.L.; Huijberts, M.S.; van Greevenbroek, M.M.; van der Kallen, C.J.; Dekker, J.M.; Nijpels, G.; Stehouwer, C.D.; et al. Plasma levels of advanced glycation endproducts nepsilon-(carboxymethyl)lysine, nepsilon-(carboxyethyl)lysine, and pentosidine are not independently associated with cardiovascular disease in individuals with or without type 2 diabetes: The hoorn and codam studies. J. Clin. Endocrinol. Metab. 2013, 98, E1369–E1373. [Google Scholar] [PubMed]
- McLellan, A.C.; Phillips, S.A.; Thornalley, P.J. The assay of s-d-lactoylglutathione in biological systems. Anal. Biochem. 1993, 211, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Nihonmatsu-Kikuchi, N.; Itokawa, M.; Rabbani, N.; Thornalley, P.J. Measurement of glyoxalase activities. Biochem. Soc. Trans. 2014, 42, 491–494. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Substrate | Product | Healthy n = 8 | EAE n = 8 | p-Value |
|---|---|---|---|---|
| Plasma | ||||
| Methylglyoxal (MGO) (nmol/L) | 3268 ± 378 | 2160 ± 230 | 0.03 | |
| Glyoxal (GO) (nmol/L) | 1414 ± 51 | 1205 ± 70 | 0.03 | |
| 3-deoxyglucosone (3DG) (nmol/L) | 2051 ± 123 | 1611 ± 152 | 0.04 | |
| Protein bound Nε-(carboxymethyl)lysine (CML) (nmol/mmol Lysine) | 25.6 ± 1 | 25.2 ± 1 | 0.83 | |
| Protein bound Nε-(1-carboxyethyl)lysine (CEL) (nmol/mmol Lysine) | 9.1 ± 0.7 | 8.4 ± 1 | 0.60 | |
| Protein bound Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine (MG-H1) (nmol/mmol Lysine) | 276.5 ± 19 | 285.6 ± 17 | 0.73 | |
| Free CML (nmol/L) | 315.4 ± 17 | 283.6 ± 15 | 0.18 | |
| Free CEL (nmol/L) | 98.1 ± 7 | 99.9 ± 9 | 0.88 | |
| Free MG-H1 (nmol/L) | 59.6 ± 6 | 91.5 ± 6 | 0.002 | |
| Spinal cord | ||||
| MGO (nmol/L) | 1980 ± 169 | 3143 ± 419 | 0.02 | |
| GO (nmol/L) | 1706 ± 250 | 2258 ± 165 | 0.09 | |
| Protein bound CML (nmol/mmol Lysine) | 297.5 ± 116 | 187.2 ± 16 | 0.36 | |
| Protein bound CEL (nmol/mmol Lysine) | 65.1 ± 16 | 37.4 ± 2 | 0.11 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 159.4 ± 10 | 165.3 ± 14 | 0.73 | |
| Free CML (nmol/L) | 61 ± 2 | 98.18 ± 8 | 0.0005 | |
| Free CEL (nmol/L) | 14.6 ± 0.4 | 18.1 ± 1 | 0.01 | |
| Free MG-H1 (nmol/L) | 3.4 ± 0.07 | 5.5 ± 0.8 | 0.01 | |
| Glo-1 activity (nmol/mg/min) | 262.5 ± 9 | 198.9 ± 14 | 0.002 | |
| Brain | ||||
| MGO (nmol/L) | 1222 ± 162 | 2302 ± 299 | 0.01 | |
| GO (nmol/L) | 1833 ± 169 | 2250 ± 89 | 0.05 | |
| Protein bound CML (nmol/mmol Lysine) | 91.5 ± 5 | 98.7 ± 6 | 0.36 | |
| Protein bound CEL (nmol/mmol Lysine) | 42.6 ± 2 | 43.7 ± 3 | 0.77 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 129.5 ± 16 | 139.4 ± 19 | 0.70 | |
| Free CML (nmol/L) | 55.6 ± 2 | 73.9 ± 3 | <0.0001 | |
| Free CEL (nmol/L) | 24.6 ± 0.8 | 25.3 ± 1 | 0.69 | |
| Free MG-H1 (nmol/L) | 3.5 ± 0.2 | 4.0 ± 0.3 | 0.19 | |
| Glo-1 activity (nmol/mg/min) | 147.9 ± 5 | 161.4 ± 3 | 0.04 |
| Substrate | Product | Vehicle n = 11 | Pyridoxamine n = 10 | p-Value |
|---|---|---|---|---|
| Plasma | ||||
| MGO (nmol/L) | 2270 ± 208 | 2488 ± 227 | 0.49 | |
| GO (nmol/L) | 1447 ± 159 | 1391 ± 91 | 0.77 | |
| 3DG (nmol/L) | 1496 ± 89 | 1642 ± 44 | 0.17 | |
| Protein bound CML (nmol/mmol Lysine) | 19.7 ± 1 | 20 ± 2 | 0.89 | |
| Protein bound CEL (nmol/mmol Lysine) | 5.3 ± 0.4 | 4.7 ± 0.4 | 0.31 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 217.0 ± 12 | 239.2 ± 15 | 0.25 | |
| Free CML (nmol/L) | 364.5 ± 137 | 218.4 ± 10 | 0.32 | |
| Free CEL (nmol/L) | 138.8 ± 70 | 71.6 ± 6 | 0.37 | |
| Free MG-H1 (nmol/L) | 96.6 ± 37 | 63.6 ± 7 | 0.42 | |
| Spinal cord | ||||
| MGO (nmol/L) | 1976 ± 177 | 1977 ± 132 | 1.00 | |
| GO (nmol/L) | 2117 ± 122 | 2207 ± 150 | 0.65 | |
| Protein bound CML (nmol/mmol Lysine) | 97.6 ± 12 | 95.9 ± 12 | 0.92 | |
| Protein bound CEL (nmol/mmol Lysine) | 50.2 ± 4 | 52.2 ± 5 | 0.75 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 108.3 ± 11 | 132.1 ± 17 | 0.25 | |
| Free CML (nmol/L) | 92.9 ± 5 | 97.3 ± 5 | 0.56 | |
| Free CEL (nmol/L) | 17.6 ± 0.4 | 17.9 ± 0.5 | 0.59 | |
| Free MG-H1 (nmol/L) | 4.5 ± 0.2 | 4.8 ± 0.3 | 0.43 | |
| Glo-1 activity (nmol/mg/min) | 1614 ± 46 | 1572 ± 59 | 0.58 |
| Substrate | Product | Wild Type n = 4 | Glo-1 Overexpression n = 5 | p-Value |
|---|---|---|---|---|
| Plasma | ||||
| MGO (nmol/L) | 1837 ± 578 | 3279 ± 432 | 0.08 | |
| GO (nmol/L) | 1047 ± 44 | 1477 ± 339 | 0.30 | |
| 3DG (nmol/L) | 1690 ± 172 | 2125 ± 167 | 0.11 | |
| Protein bound CML (nmol/mmol Lysine) | 25.9 ± 0. 8 | 26.9 ± 2 | 0.64 | |
| Protein bound CEL (nmol/mmol Lysine) | 6.2 ± 1 | 8.8 ± 0.8 | 0.08 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 307.9 ± 51 | 274.0 ± 15 | 0.50 | |
| Free CML (nmol/L) | 251.1 ± 28 | 790.4 ± 523 | 0.39 | |
| Free CEL (nmol/L) | 72.7 ± 15 | 330.4 ± 250 | 0.39 | |
| Free MG-H1 (nmol/L) | 64.0 ± 8 | 169.1 ± 114 | 0.44 | |
| Spinal cord | ||||
| MGO (nmol/L) | 3865 ± 500 | 3382 ± 184 | 0.35 | |
| GO (nmol/L) | 2794 ± 630 | 2517 ± 220 | 0.66 | |
| Protein bound CML (nmol/mmol Lysine) | 108.4 ± 4 | 102.3 ± 8 | 0.54 | |
| Protein bound CEL (nmol/mmol Lysine) | 41.9 ± 6 | 46.5 ± 9 | 0.69 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 270.7 ± 25 | 305.4 ± 47 | 0.57 | |
| Free CML (nmol/L) | 87.3 ± 9 | 92.9 ± 7 | 0.64 | |
| Free CEL (nmol/L) | 17.1 ± 0.8 | 16.4 ± 0.5 | 0.53 | |
| Free MG-H1 (nmol/L) | 4.4 ± 0. 8 | 3.7 ± 0.1 | 0.32 | |
| Glo-1 activity (nmol/mg/min) | 66.6 ± 8 | 367.8 ± 6 | <0.0001 | |
| Brain | ||||
| MGO (nmol/L) | 2906 ± 309 | 2836 ± 127 | 0.83 | |
| GO (nmol/L) | 4086 ± 718 | 4291 ± 354 | 0.79 | |
| Protein bound CML (nmol/mmol Lysine) | 63.9 ± 4 | 57.8 ± 7 | 0.50 | |
| Protein bound CEL (nmol/mmol Lysine) | 40.6 ± 6 | 37.0 ± 2 | 0.55 | |
| Protein bound MG-H1 (nmol/mmol Lysine) | 177.3 ± 16 | 179.4 ± 13 | 0.92 | |
| Free CML (nmol/L) | 52.0 ± 5 | 51.2 ± 3 | 0.88 | |
| Free CEL (nmol/L) | 25.4 ± 2 | 25.4 ± 1 | 0.97 | |
| Free MG-H1 (nmol/L) | 3.3 ± 0.1 | 3.4 ± 0.2 | 0.69 | |
| Glo-1 activity (nmol/mg/min) | 50.1 ± 2 | 296.1 ± 11 | <0.0001 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wetzels, S.; Wouters, K.; Miyata, T.; Scheijen, J.L.J.M.; Hendriks, J.J.A.; Schalkwijk, C.G.; Vanmierlo, T. Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression. Int. J. Mol. Sci. 2018, 19, 1311. https://doi.org/10.3390/ijms19051311
Wetzels S, Wouters K, Miyata T, Scheijen JLJM, Hendriks JJA, Schalkwijk CG, Vanmierlo T. Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression. International Journal of Molecular Sciences. 2018; 19(5):1311. https://doi.org/10.3390/ijms19051311
Chicago/Turabian StyleWetzels, Suzan, Kristiaan Wouters, Toshio Miyata, Jean L. J. M. Scheijen, Jerome J. A. Hendriks, Casper G. Schalkwijk, and Tim Vanmierlo. 2018. "Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression" International Journal of Molecular Sciences 19, no. 5: 1311. https://doi.org/10.3390/ijms19051311
APA StyleWetzels, S., Wouters, K., Miyata, T., Scheijen, J. L. J. M., Hendriks, J. J. A., Schalkwijk, C. G., & Vanmierlo, T. (2018). Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression. International Journal of Molecular Sciences, 19(5), 1311. https://doi.org/10.3390/ijms19051311